第五章
课程来源与翻译声明
- 原著版权:本教程翻译自瓦赫宁根大学(Wageningen University & Research)的经典开源课程 Introduction to Bioinformatics。
- 翻译说明:本版本主要采用人工智能(机器翻译)进行全文本地化,并由团队进行了初步校对。部分专业术语可能存在翻译不够地道或准确的地方。
- 勘误反馈:若你在学习过程中遇到语病、错别字、代码失效或概念歧义,欢迎随时点击页面右上角的 :material-edit: 编辑此页(或联系课题组/在 GitHub Issue 中)提交反馈,帮助我们持续完善本教程。
本章将学习多种高通量生物分子测量技术。
学习目标
学习本章后,你应当能够: - 解释组学测量和系统生物学的意义 - 讨论 DNA 和 RNA 测序以及质谱测量的核心原理及其数据的局限性 - 列举主要的定量数据分析技术(聚类、分类、差异表达)并描述其用途 - 描述测序数据在基因组和转录组分析中的应用
关于 -omics 的词源
词源(来自维基百科)
-ome("某一类别的整体")+ -ics,两者均经由国际科学词汇和新拉丁语最终源自古希腊语
后缀
-omics(主要用于生物学领域)
构成名词,意为"对某事物整体的研究"。
本章讨论我们所说的组学测量:基因组学、转录组学(基因表达)、蛋白质组学和代谢组学。组学技术测量细胞中不同类型分子的存在、水平和/或相互作用,一次性获取所有分子的数据(见图:不同组学层级)。基因组学侧重于可从基因组中获得的所有信息(结构、功能、进化等)。转录组学、蛋白质组学和代谢组学分别侧重于基因表达、蛋白质和代谢物的水平。最后,表型组学测量细胞和生物体的外观和行为。
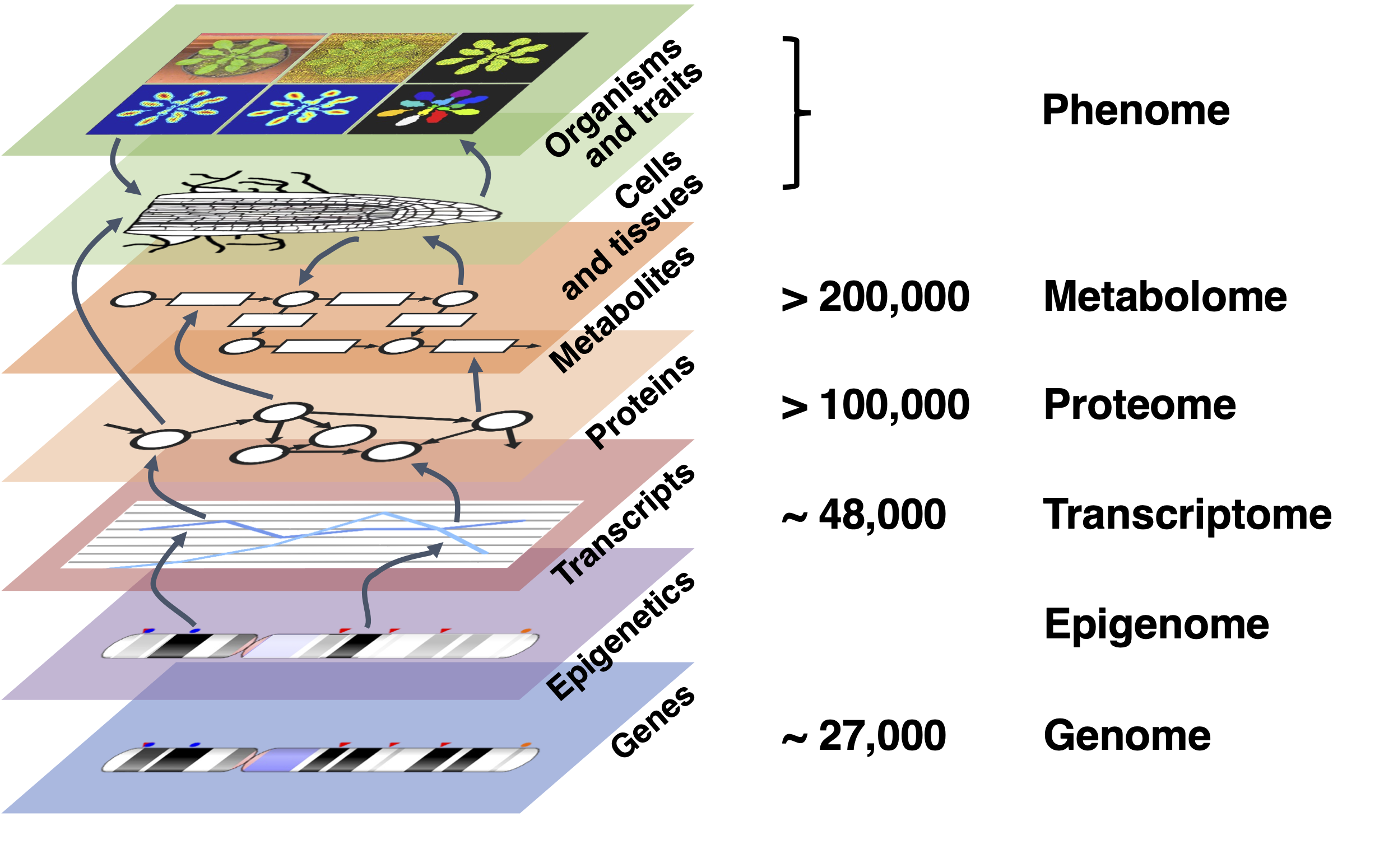
图:不同的组学层级,以 拟南芥 (Arabidopsis thaliana) 的估计数量为例进行说明。基因在表观遗传调控的影响下表达为转录本。转录本被翻译为蛋白质,在细胞中执行多种功能,其中包括对消耗和产生化合物的代谢反应的酶促调控。蛋白质和代谢物调控参与组织生长和发育的细胞间相互作用,最终形成生物体和可观察的性状。在每个层级,现在都有手段在非常广泛的、所谓的"基因组范围"尺度上测量分子的存在/缺失、水平和某些类型的相互作用。图片来源:CC BY-NC 4.0 [@own_5_2024]
基因的概念是分子生物学中心法则的核心;因此早期大量研究投入到基因组测序中是理所当然的。随后这些基因组被注释基因,并预测了相应的蛋白质序列。这种对序列的关注主导了生物信息学最初几十年的发展,推动了前面章节中讨论的序列比对、系统发育和基于序列的结构预测等数据库和工具的开发。然而,在测序第一批基因组后人们发现,DNA 只讲述了整个故事的一部分:基因和蛋白质的表达及其在细胞内和细胞间过程中的相互作用决定了细胞和生物体的行为。这促成了功能基因组学和系统生物学的研究,其中对其他组学层面数据的计算分析变得不可或缺。
下面将首先介绍基因组学以及最相关的技术——测序(该技术也用于转录组学)。然后介绍功能基因组学和系统生物学,并对转录组学、蛋白质组学、代谢组学和表型组学以及涉及的主要数据分析类型进行简要概述。
基因组学与测序¶
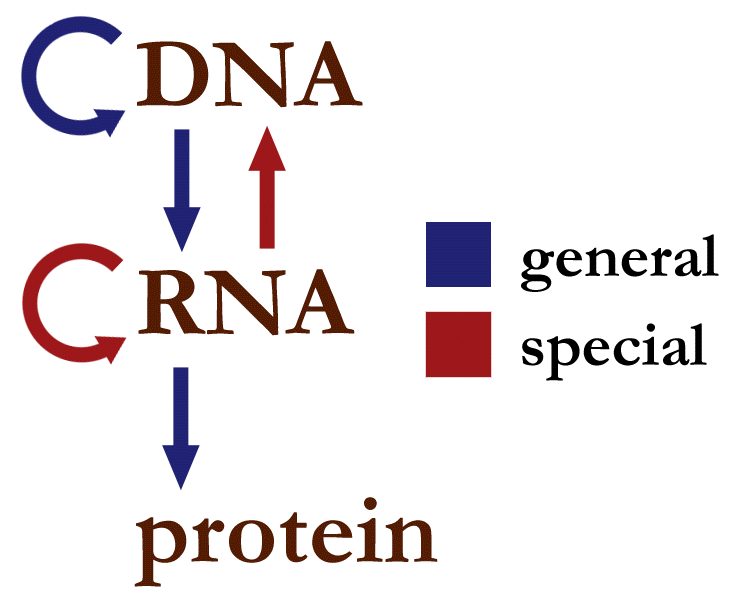
图:细胞中的信息流。图片来源:CC0 1.0 [@central_dogma_2008]
DNA 是生物信息传递链的起点(中心法则)。从 DNA 出发,经过转录和翻译,最终形成完整的生物体及其表型。因此,从起点开始讨论是合理的。甚至在人们了解 DNA 及其作为遗传信息载体的作用之前,就已经知道亲本的特征会遗传给后代。大约在 1866 年,孟德尔 (Gregor Mendel) 是第一个进行详细实验测试遗传性的人。他首先描述了"遗传单位",后来被命名为基因。今天,我们知道基因编码在我们细胞的 DNA 中。我们对基因的理解已经扩展为一个更复杂的概念,集中在编码蛋白质或 RNA 的 DNA 片段上。"基因组"一词最初用于描述一个生物体或细胞中的所有基因,但现在指的是一个细胞的全部 DNA 含量。
知识框 5.1:DNA 测序的历史与人类基因组计划
基因组测序的历史与人类基因组计划(HGP)密切相关,而 HGP 是技术发展的主要驱动力。一切始于 1977 年 Sanger、Maxam 和 Gilbert 开发的第一种 DNA 测序方法,这使得第一个基因组——Phi X 噬菌体得以被测序。
另一个重要里程碑是 1985 年 PCR(聚合酶链式反应,见第 2 章)的发展,它实现了 DNA 扩增。第一台自动化测序仪(AB370A)于 1986 年问世。
随着技术的不断发展,新的基因组序列开始缓慢出现。EB 病毒于 1983 年被测序。第一个细菌基因组——流感嗜血杆菌 (Haemophilus influenzae) 于 1995 年被测序,随后是第一个古菌基因组——詹氏甲烷球菌 (Methanococcus jannaschii) 于 1996 年被测序。同一年,第一个真核生物基因组——酿酒酵母 (Saccharomyces cerevisiae) 也被测序。主要细菌模式生物大肠杆菌 (Escherichia coli) 于 1997 年被测序。
人类基因组计划于 1988 年正式启动,尽管测序工作直到约 1990 年才开始。人类基因组使用 Sanger 测序技术(下文将描述)完全测序,覆盖估计 32 亿碱基对中 90% 的初稿基因组于 2001 年发表在 Nature 上,最终基因组于 2003 年完成。最令人惊讶的发现之一是人类基因组仅包含约 20,000 个基因,远低于估计的 50,000-140,000 个。整个项目的成本估计为 $30 亿。2021 年,使用下文描述的第三代技术——PacBio 和 Oxford Nanopore,完成了人类基因组的首个真正的端粒到端粒组装。
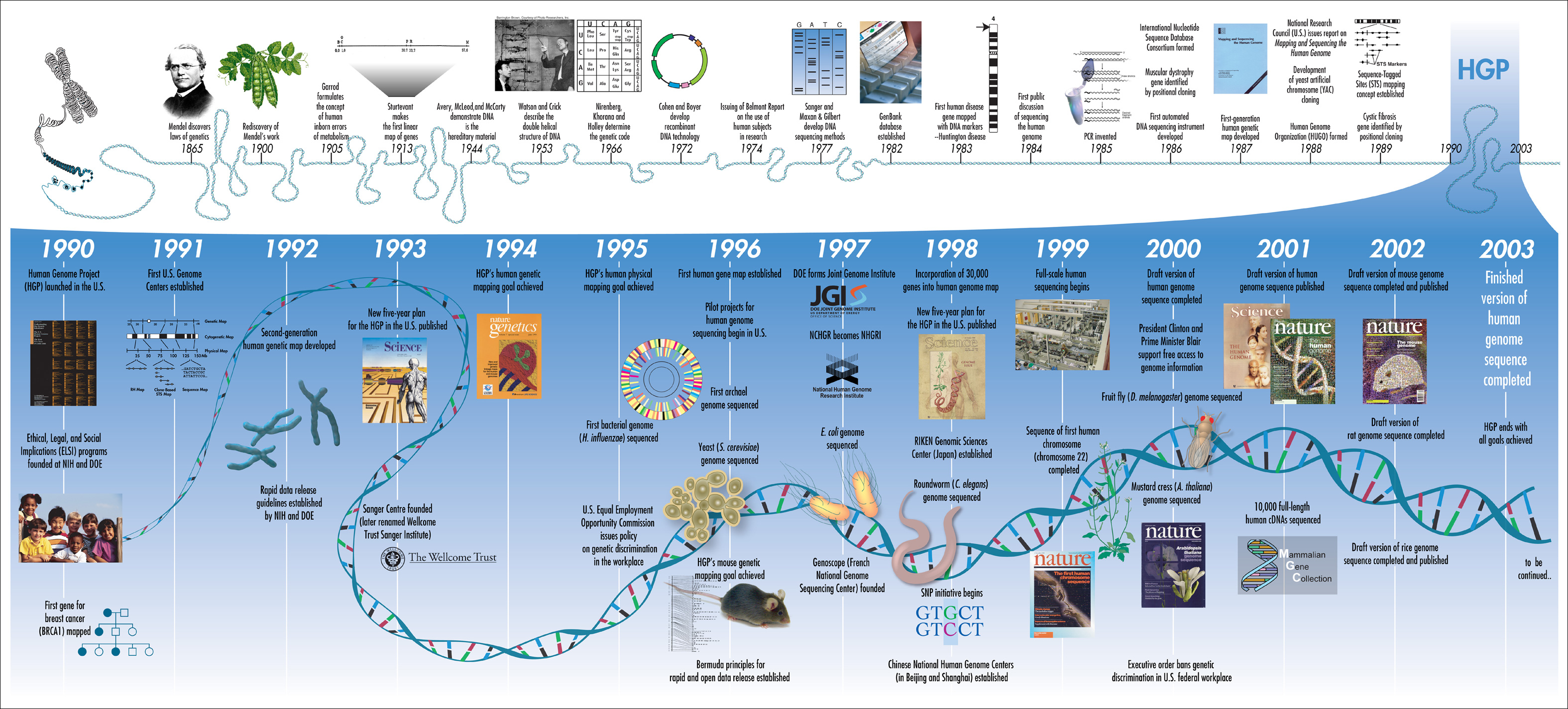
图:人类基因组计划时间线。图片来源:CC-BY 2.0 [@timeline_HGP_2003]
基因组¶
基因组测序的历史以及人类基因组计划在测序方法发展中的重要性在知识框 5.1 中有描述。随着测序技术的快速发展,我们对基因组及其内容的理解也在不断增长。我们现在知道基因组在大小、染色体数量和倍性方面差异巨大(见图:基因组大小与倍性),基因含量也各不相同(见下表)。基因组大小从细菌的 100kb 到植物中超过 100Gb 不等。人类的基因组大小为 3.2Gb。
| 物种 | 基因组大小 (kb) | 基因数量 | 转录本数量 | 平均基因密度 (kb) |
|---|---|---|---|---|
| E. coli | 4.6 | 4288 | 4688 | 1.1 |
| S. cerevisiae | 12.1 | 6600 | 7127 | 1.8 |
| C. elegans | 100 | 19985 | 60000 | 5 |
| D. melanogaster | 143.7 | 13986 | 41620 | 10.2 |
| A. thaliana | 119.1 | 27562 | 54013 | 4.3 |
| H. sapiens | 3100 | 20077 | 58360 | 154.4 |
表:模式物种的基因组大小和基因数量
不仅基因组大小在不同生物体之间差异巨大,在真核生物中染色体数量和染色体拷贝数(倍性)也是如此。染色体数量范围从果蝇 (Drosophila) 的 4 条到人类的 23 条,再到金鱼的 50 条和某些蕨类植物的 100 条以上。同样,倍性范围从单倍体(单套染色体,倍性为 N)和二倍体(两份拷贝,倍性为 2N)到多倍体(超过 3 份拷贝),极端情况下蕨类植物的倍性水平超过 100。基因数量也因物种而异;在低端,细菌内共生体有 120 多个基因,而大多数高等真核生物(包括人类)有 15,000 到 25,000 个基因,一些植物可以有超过 40,000 个基因——水稻有超过 46,000 个基因。
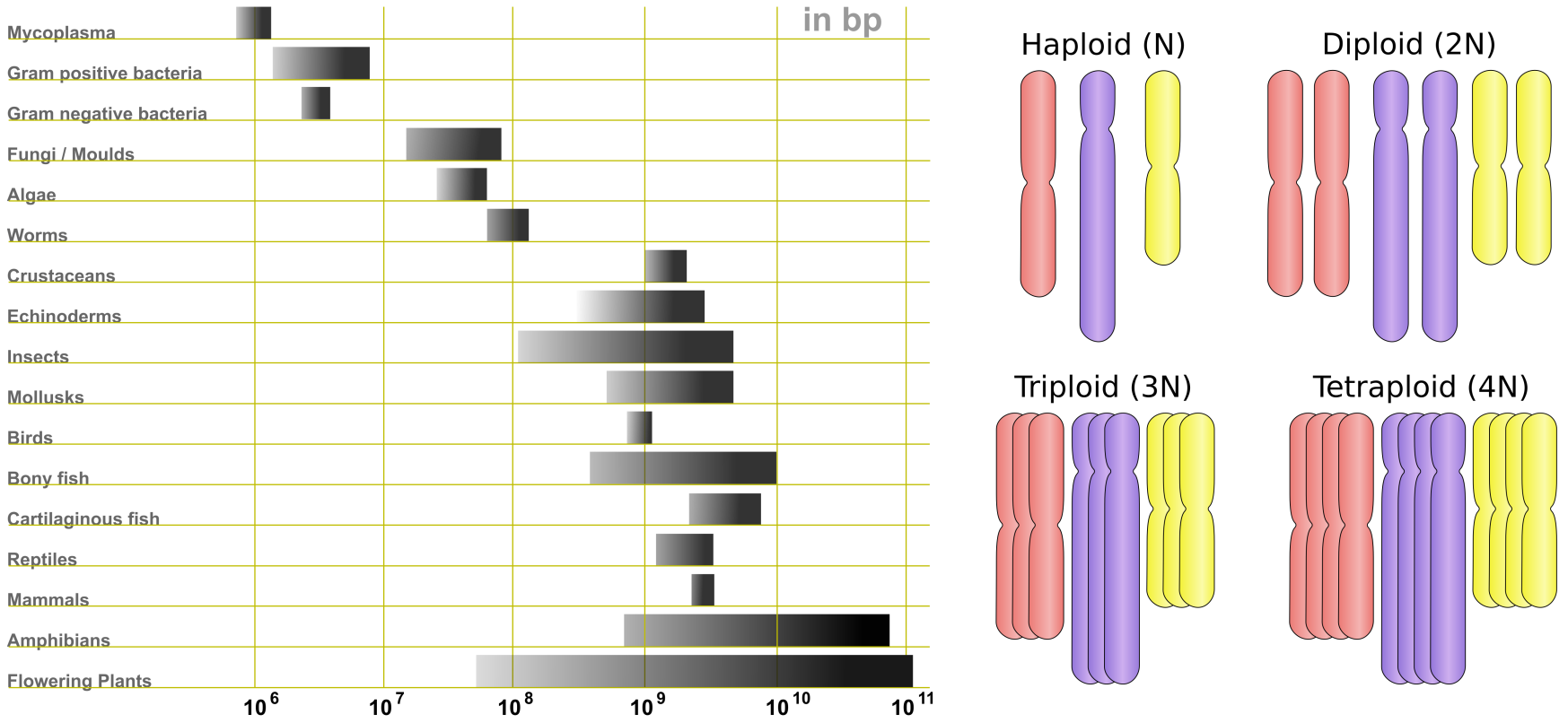
图:左图为各种基因组大小。图片来源:CC BY-SA 3.0 [@gene_ploidy_2010];右图为倍性示例。图片来源:CC BY-SA 3.0 [@gene_ploidy_2011]
知识框 5.2:大小有(没)关系?
基因组有各种形状和大小。已知最小的(非病毒)基因组是细菌内共生体 Nasuia deltocephalinicola 的基因组,仅由 112,091 个核苷酸组成,编码 137 个蛋白质。迄今为止已知最大的基因组是大理石肺鱼和植物 Fritillaria assyriaca,含有 130,000,000,000 个核苷酸(130Gb),尽管据说变形虫 Polychaos dubium 的基因组大小为 670Gb。基因组大小不一定与基因组中的基因数量相关(另见上表)。反过来,蛋白质编码基因的数量也并不总是随着生物体的复杂性而增加。
基因组测序技术¶
为了研究基因组,我们需要一种人类可读的表示方式。这需要"读取"DNA 分子为 A、C、T 和 G。产生基因组的过程始于 DNA 测序,即检测核苷酸及其在 DNA 链上的排列顺序。
如果你使用基因组数据,对所用技术及其优缺点有基本了解非常重要。这使我们能更好地理解所处理数据的质量以及可以从中获得哪些生物学见解。
从概念上讲,有三种测序方式:
-
**化学测序**依赖于从链上逐步切下最后一个核苷酸并鉴定它。由于使用放射性标记,这种方法从未被广泛使用。
-
**边合成边测序**涉及逐个碱基合成互补链并检测每个位置的插入。这是目前最广泛使用的方法,有多种实现方式。它也被称为下一代测序(NGS)。
-
**直接测序**涉及直接测量 DNA 链中核苷酸的顺序,迄今为止仅在 Oxford Nanopore 测序 中实现。
不同技术在产生的 DNA 序列(即测序读段)长度和通量方面差异很大,它们共同决定了覆盖度:基因组中每个碱基被读段代表的(平均)次数。对于某些目的(如基因组组装),覆盖度必须足够高——根据读段长度,通常在 50x 到 100x 之间。下图展示了若干测序设备及其在每次运行中的读段长度和产量方面的能力。
测序技术在碱基识别(某位置检测到的核苷酸)的准确性方面也有所不同。这种准确性用质量分数或 Q 分数来衡量,它们代表碱基识别错误的概率。Q 分数越高越好。最常用的阈值是 Q30,对应于错误碱基识别概率为千分之一,即准确率为 99.9%。
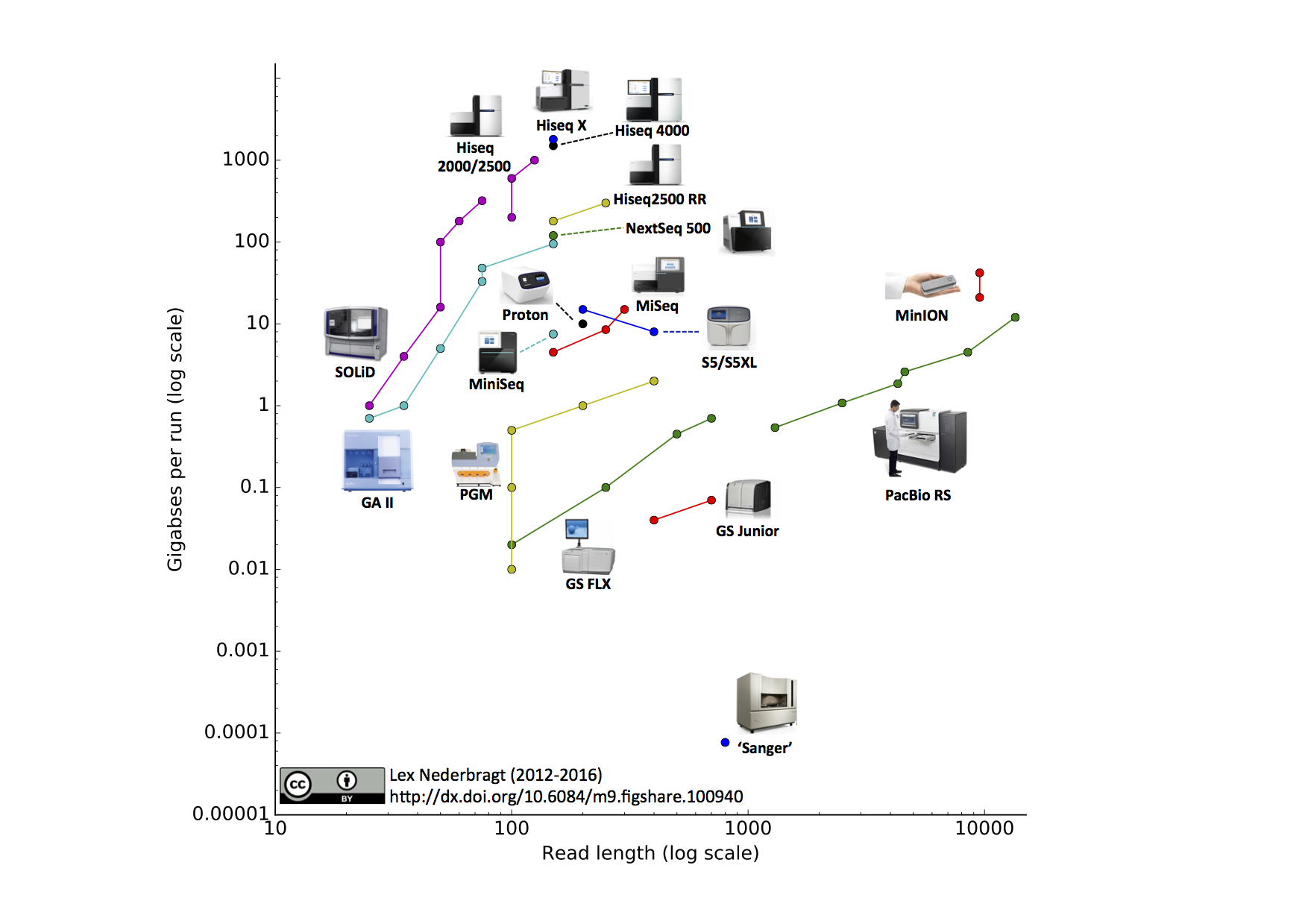
图:测序技术,产量与读段长度的关系。注意对数坐标。每种技术的多个点表示因升级带来的读段长度和/或产量的改进。该图已经过时,Illumina NovaSeq 产生了更高的产量,Oxford Nanopore MinION/PromethION 和 PacBio Sequel II 设备产生了更长的读段。图片来源:CC BY 4.0 [@sequencing_technology_2016]
Sanger 测序¶
Sanger 测序是第一种"高通量"DNA 测序方法。关于其历史和工作原理的更多细节,请参见知识框 5.8。
本质上,Sanger 测序是一种 PCR 反应,使用不同颜色的荧光核苷酸逐步创建 DNA 片段的拷贝,通过检测荧光信号并将其转化为核苷酸。Sanger 测序需要在测序反应中存在许多单一、独特的 DNA 片段的拷贝,以确保光信号足够强以被检测。每一步的信号还必须是单一颜色的,因此模板序列必须完全相同。这意味着在样品准备步骤中,片段通过普通 PCR 或其他大规模扩增方法进行克隆。对唯一性的要求是 Sanger 测序最大的缺点。
关于 Sanger 测序的重要知识
- 它是最初的测序平台。
- 被用于测序第一个人类基因组。
- 它产生长达 1000bp 的读段,质量为 99.9%(Q30)。
- 它是一种低通量方法。
- 它一次只能测序一个片段。
Sanger 测序一直是最主要的测序平台,直到约 2007 年。从 2004 年起,它越来越多地被所谓的下一代测序(NGS)方法所取代。今天它仍被使用,其中包括验证变异的 PCR 产物测序、确定克隆载体中基因的方向,或在微卫星研究中使用。
下一代测序¶
下一代测序(NGS)技术允许以远低于 Sanger 测序的成本实现更高的通量,但代价是:更短的读段和更低的碱基识别准确率。这些较新的设备现在每次测序运行可产生数十亿条读段。详细介绍所有这些方法超出了本课程的范围。
与 Sanger 测序一样,NGS 方法依赖于对输入 DNA 片段文库的扩增,以增强实际测序步骤的信号。目前大多数测序数据由 Illumina 技术(或第三代方法,见下一节)产生,该技术允许大规模并行测序读段。边合成边测序和 Illumina 图形化流动池的工作原理在知识框 5.9 中有详细解释;下面我们列出数据的主要特征。
关于 Illumina 测序的重要知识
- 一次运行中所有读段长度相同,由循环数决定(20-350bp)。
- 由于固定的读段长度,测序读段可能包含引物序列(如果 DNA 片段短于循环数);这些必须被去除。
- 读段可以只从一个引物端测序,产生所谓的单端读段,或从两个引物端测序,产生双端读段,即来自同一分子的两条读段,它们之间的距离大致已知。
- 读段的碱基准确率约为 99.99%(Q40)。
- 通量非常高:单次运行获得的读段数量从数百万到数十亿不等,取决于设备型号。
- 具有极端 GC 含量的片段不太容易被测序,这可能导致基因组组装不完整或覆盖度不均匀。
- 适用于变异检测(见变异检测)。
总体而言,Illumina 读段成本低、读长短且准确率极高。
第三代测序¶
在 NGS 成功之后,替代性的所谓第三代技术被引入以克服一些不足,主要是有限的读段长度。所有这些方法都产生更长的读段,但产量较低,且错误率通常略高于之前描述的方法。它们还执行所谓的实时测序:每个 DNA 片段以连续方式完全测序,不需要像 NGS 那样的独立循环。
PacBio¶
最成熟的方法是 PacBio 单分子实时(SMRT)测序。与其他方法相比,它不包含 PCR 步骤来扩增模板 DNA 的信号。相反,在测序单个 DNA 分子时,当标记的核苷酸被插入时会发出光信号。该光信号随后被放大以便检测。更多细节请参见知识框 5.10。
关于 PacBio 测序的重要知识
- 具有极端 GC 含量的片段可以被测序,因为没有 PCR 步骤。
- 单个 DNA 片段被实时测序。
- 同一片段可以被多次测序并用于纠错(HiFi)。
- HiFi 读段长度约为 15kb,连续长读段(CLR)最长可达 175kb。
- 准确率从 CLR 的 99%(Q20)到 HiFi 读段的 99.9%(Q30)不等。
- 它是一种高通量技术,一次运行可产生多达 2500 万条读段(Revio)。
- 成本仍高于 Illumina。
- 适用于基因组组装。
Nanopore 测序¶
最新的技术是纳米孔测序,目前由 Oxford Nanopore 在 MinION 及相关设备上提供(见图)。该技术与其他所有技术完全不同,因为它直接基于 DNA 单链被拉过嵌入在膜中的蛋白质纳米孔时引起的电流变化来检测核苷酸顺序(更多信息见知识框 5.11)。与 PacBio 测序一样,读段长度由 DNA 模板的长度决定。

图:Oxford Nanopore MinION 测序仪。图片来源:CC BY-SA 4.0 [@minion_2020]
关于纳米孔测序的重要知识
- 它可以测序非常长的读段。
- 准确率为 96.8-99.9%(Q15-Q30)。
- 它可以直接检测碱基修饰(甲基化)。
- 具有极端 GC 含量的片段可以被测序,因为没有 PCR 步骤。
- 单个 DNA 片段被逐一测序,实现实时测序。
- 它是一种高通量技术,每个流动池产生 250 万到 350 万条读段(MinION)。
- 适用于基因组组装。
质量控制¶
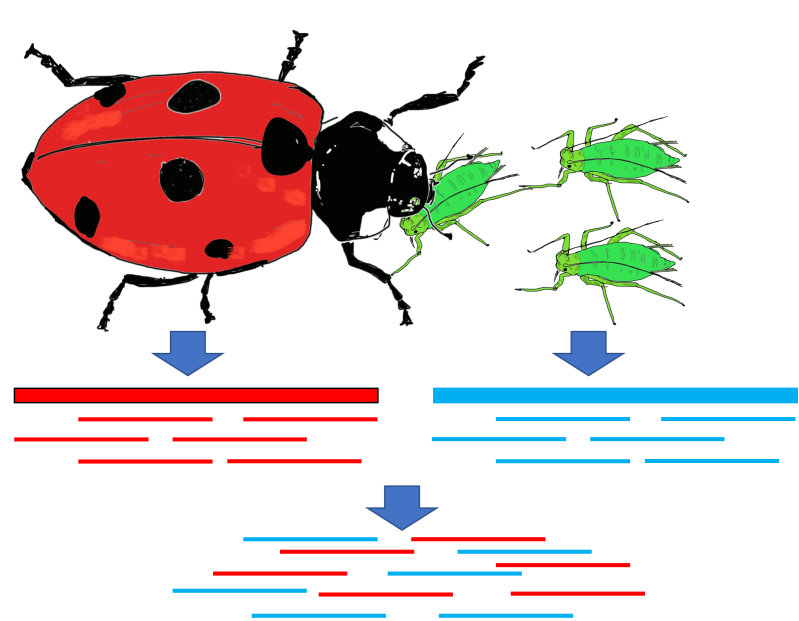
图:测序样品污染的原因,以瓢虫及其主要食物来源蚜虫为例。此外,所有真核生物都有一个由原核生物、病毒和小型真核生物组成的微生物组,这些也可能作为污染物存在。图片来源:CC BY-NC 4.0 [@own_5_2024]
在将测序数据用于进一步分析之前,我们需要确保数据质量足够好。
质量控制的第一步是检查数据的准确性。由于测序技术并不完美,输出中必然存在错误。为了尽量减少这些错误,我们去除低质量读段或碱基。
与测序本身相关的错误是碱基识别错误(替换错误)、未识别碱基(插入/缺失)、GC 偏差、同聚物、读段 3' 端质量下降和重复(扩增偏差)的结果。
有时候我们测序的内容是正确的,但不是我们最初想要测序的内容(见图)。识别污染对于基因组组装很重要,因为我们不想将基因组序列错误地分配给错误的物种。想象一下,如果瓢虫突然拥有了红色蚜虫眼睛的基因会造成的混乱。已有工具利用序列同源性(例如 BLAST)来识别来自污染物物种的读段。
除了输入样品的污染外,测序数据还可能含有接头和测序载体的残留,这些可以用专用软件去除。
在进一步分析之前,评估测序本身的质量和输出数据非常重要。
基因组组装¶
当某个物种没有参考基因组时,我们需要从头组装一个,即将 DNA 序列读段拼接在一起构建基因组。这里讨论步骤和注意事项:首先,我们探讨为什么要创建参考组装以及可以创建哪些类型的参考。然后介绍组装过程及其挑战。最后讨论基因组注释和结构变异的检测。
参考基因组质量¶
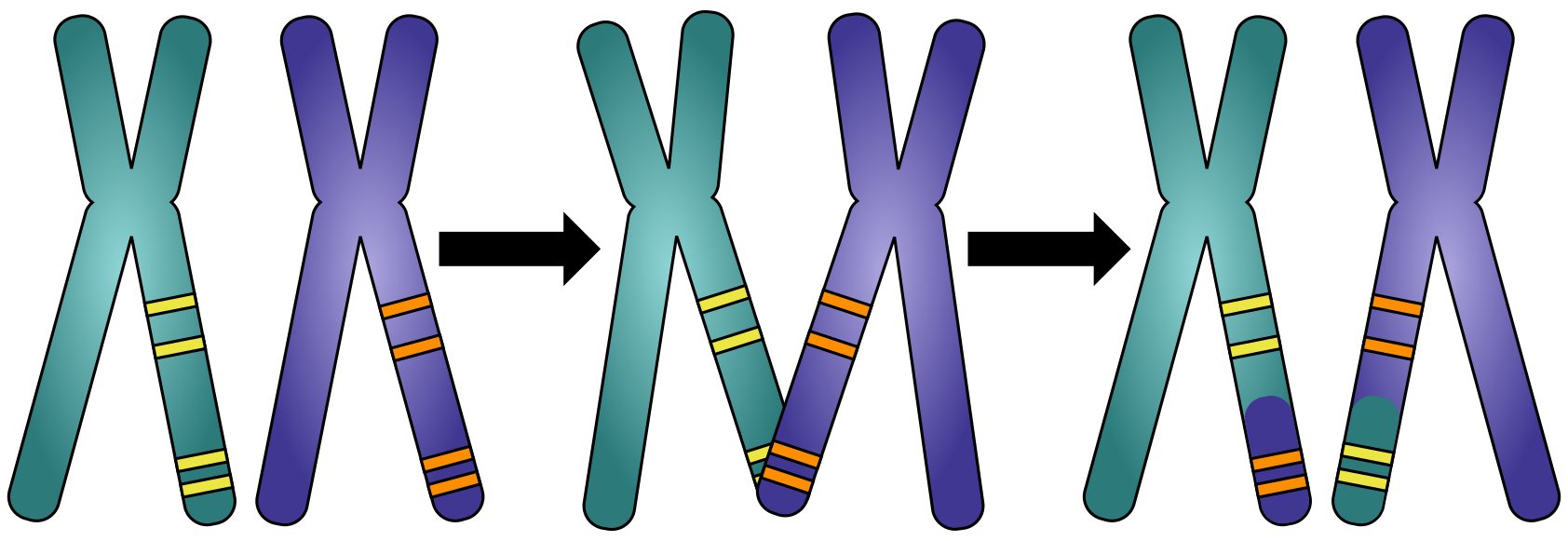
图:等位基因的共分离:基因组的哪些部分是从母亲或父亲一起遗传的。图片来源:CC BY-NC 4.0 [@own_5_2024]
基因组可以有不同的重建目标,这会影响最终组装所需的质量。例如,人类基因组已经尽可能地被组装,2021 年发表了第一个端粒到端粒组装,增加了最后 5% 的碱基。达到这个阶段付出了巨大的努力,无论是在资金还是人力方面。对于每个基因组组装项目来说,这既不可行也并非严格必要。因此,目前大多数可用的基因组组装是所谓的草图组装,大多数完全完成的基因组来自细菌和其他小基因组物种。在组装过程方面,对于真核生物基因组,常染色质区域组装效果最好。幸运的是,这些区域包含大多数基因,使草图组装可用于研究突变或表达模式。然而,当我们想要研究基因组本身的大规模特征(如等位基因共分离或基因顺序)时,需要更连续的组装。第三代测序、现代组装技术和其他新技术使染色体水平的组装越来越容易实现。
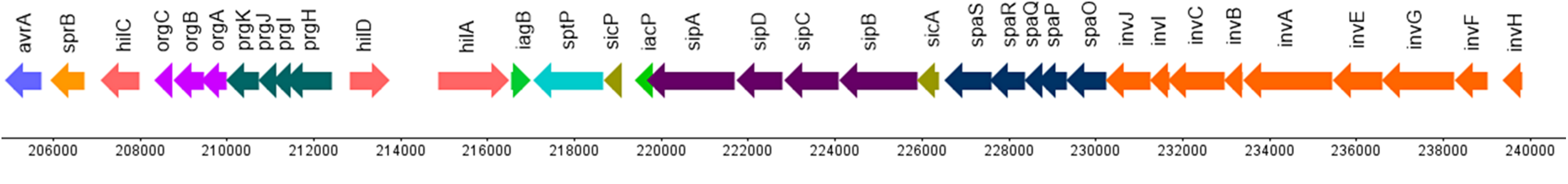
图:肠道沙门氏菌肠道亚种 斯坦利血清型致病岛-1 的遗传表示。箭头表示基因及其方向,x 轴为基因组坐标。图片来源:CC BY 4.0 修改自 [@salmonella_alt_2019]
基因组组装策略¶
在 DNA 测序的早期,生成测序读段成本高且速度慢。因此大量精力投入到开发需要最少序列数据即可组装基因组的方法。待测序的基因组部分被克隆到细菌载体中并以此方式扩增。这些大片段(远长于读段长度)然后从一端开始测序。接着,从已测序部分的末端生成新的测序引物用于下一轮测序,直到片段末端。这种相当繁琐的方法对于较大的基因组不可行。这导致了全基因组鸟枪法测序方法的开发,该方法的实现在于计算能力的增长。在这种方法中,克隆的 DNA 片段被打碎成更小的片段并一次性全部测序。随着第二代测序的出现,这种方法通过省略克隆步骤进行了更新。第二代测序技术(如 Illumina)允许同时测序混合片段,产生的测序数据量巨大。因此,不再需要实验工作来选择测序哪一部分,而是将所有内容一次性测序,然后在计算上解决这个拼图。
全基因组测序¶
如今,最广泛使用的基因组测序方法是全基因组测序(WGS)。顾名思义,整个基因组被测序,不做区分。从细胞中提取 DNA 并随机打碎成片段。这些片段经过大小选择后,使用 Illumina、PacBio 或 Oxford Nanopore 技术(或其组合)进行测序。需要注意的是,对于较大的真核生物基因组,WGS 通常生成草图基因组组装;需要额外步骤才能获得完整的、高质量的参考基因组组装。另一方面,来自长读段的细菌组装通常是完整的。
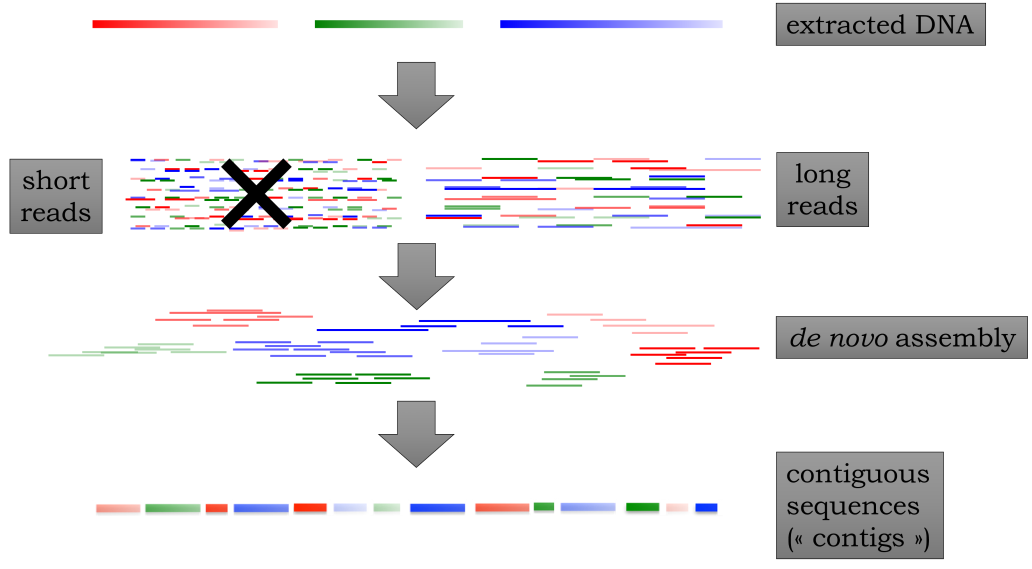
图:全基因组测序与组装。左侧的短读段通常不再用于从头组装(原核生物和病毒除外)。同时,许多可用的参考基因组也是这样产生的。Contigs(重叠群)是指可以无歧义组装的最长基因组片段(另见重复序列)。图片来源:CC BY-NC 4.0 [@own_5_2024]
组装挑战¶
组装的主要挑战是从数百万或数十亿个小读段中重建原始基因组序列。有些人将这个过程比作将一堆报纸(每份报纸代表基因组的一个拷贝)放入碎纸机,然后试图从碎纸屑中重建一份原始报纸。解决这个拼图推动了专用组装算法和软件的发展。任何计算方法都必须克服测序数据和基因组特征所带来的现实挑战。
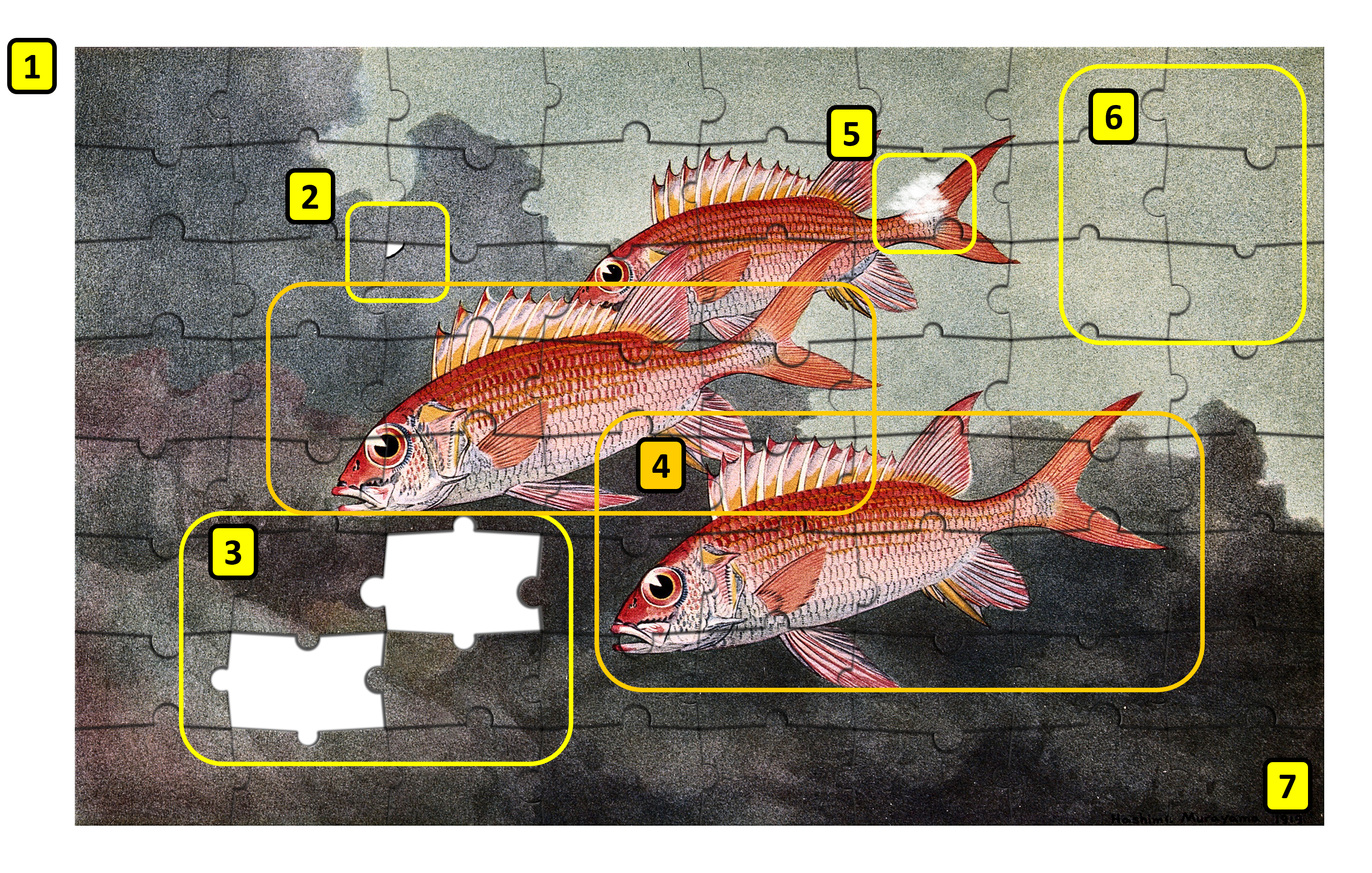
图:将组装问题类比为拼图游戏。编号在下方文本中被引用。图片来源:基于 Public Domain Mark [@three_fish],CC BY-NC 4.0 [@own_5_2024]
如果我们用拼图类比来看基因组组装(见图),挑战就变得明显了:
-
拼图盒上没有图片,即我们不知道组装后的基因组应该是什么样的。我们可以查看相关基因组,但这只能给出一个大致的概念(图中的编号 1)。
-
拼图中有大量的拼块,数十亿块。每一块代表基因组中被测序的一小部分。
-
有些拼块边缘破损或有污渍,即读段包含错误,进一步模糊了整体画面(编号 2 和 5)。
-
有些拼块丢失了。基因组的某些部分不像其他部分那样容易被打碎,因此不包含在打断的片段中。其他部分具有极端的 GC 值,测序效率较低(编号 3)。
-
拼图的某些部分包含相同的图像。用基因组的术语来说,这些是重复区域,其中某些基因可能有一个以上的拷贝。例如,核糖体 RNA 操纵子(编码核糖体各部分区域)由多个拷贝组成(编号 4)。
-
拼图的某些部分看起来完全相同且没有特征:重复区域(编号 6)。
-
在环形基因组中,没有"角落":我们不知道基因组从哪里开始或结束(编号 7)。
除了单个拼图的类比之外,许多生物体包含两份(即二倍体)或更多(即多倍体)相同的染色体拷贝,它们之间存在微小差异。本质上,在这种情况下我们试图从两个(或更多)略有不同版本的拼图中组装一个拼图。如果这些差异太大,来自两个拼图的部分可能会被独立组装而不被注意到(记住——我们没有拼图盒上的图片!)。
使用长的高质量读段,这个拼图挑战变得更加简单,因为总拼块更少,无特征的片段也更少。目前,使用 PacBio HiFi 读段结合其他技术可以常规生成染色体水平的组装。
重复序列¶
主要是真核生物基因组含有沿基因组有许多近乎相同拷贝的序列。这些重复区域(在拼图类比中是背景)是基因组组装的主要挑战,大多数重叠群(contigs,连续序列,可以无歧义组装的最长片段)在重复区域的边缘终止。鉴于重复序列近乎相同的性质,组装软件很难确定哪个读段属于哪个重复元件拷贝。所使用的证据位于重复序列的边缘,在那里它与非重复序列重叠。这个过程就像找到许多包含鱼和背景碎片的拼图块,并试图弄清楚哪条边属于哪条鱼以及中间有多少背景。解决重复问题的一个方案是更长的读段(可以桥接两条鱼之间的"海域")。为了说明重复在组装中造成的问题规模:大多数哺乳动物的 Y 染色体有一半以上尚未被组装,因为重复含量。第 1 章介绍了如何在基因组注释之前作为第一步屏蔽基因组中的重复序列。
组装质量评估¶
在组装新基因组时,我们无法通过与已知真实值进行比较来验证其质量。我们也很少能得到完整的基因组或染色体作为单个重叠群的结果。因此,需要其他指标和方法来评估组装质量。我们可以利用在其他组装项目中积累的经验来评估一个组装的质量;我们可以将其与已经组装好的密切相关的基因组进行比较;我们还可以根据物种的已知生物学知识,将组装与我们对基因组在总体大小、染色体和基因数量方面的预期进行比较。
结构与功能注释¶
基因组组装之后的下一步是注释。首先,在结构注释中我们试图识别(蛋白质编码)基因的位置和结构(另见第 1 章 - 基因预测)。然后,在功能注释中我们尝试识别预测基因的功能(另见第 1 章 - 功能注释)。
完整基因组的见解¶
组装的连续性和完整性决定了我们能从中获得什么信息。在参考基因组质量一节中已经提到了共分离等位基因和基因顺序。如果一个基因组被组装成更少、更大的片段(即更长的重叠群),我们还可以更多地了解在基因表达调控中发挥作用的长距离调控元件(见下图)。
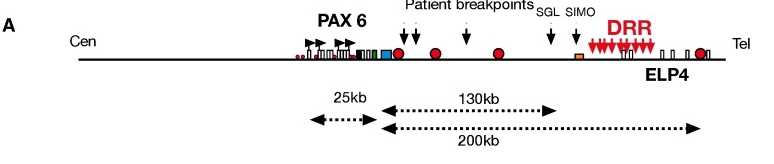
图:人类 PAX6 位点的物理图谱,显示长距离调控元件。DRR 区域调控 PAX6 的表达,红点显示调控元件的确切位置。虚线箭头表示这些调控元件与 PAX6 之间以千碱基为单位的距离。图片来源:[@pax_locus]
如上所述,人类基因组的端粒到端粒组装增加了之前缺失的 5% 的基因组序列。虽然之前的人类基因组组装已被认为是黄金标准并且非常完整,但基因数量增加了 5%,其中 0.4% 为蛋白质编码基因。已识别基因的增加也使得研究这些基因的表达模式成为可能。基因组覆盖率的增加还可以揭示隐藏的元件。作为示例,下图展示了一个疾病相关基因的所有旁系同源物,它们最终被解析出来。大多数缺失的拷贝位于基因组中难以测序的部分。染色体水平的组装还使我们能够研究基因组进化本身——染色体在进化和物种形成过程中如何重新排列。
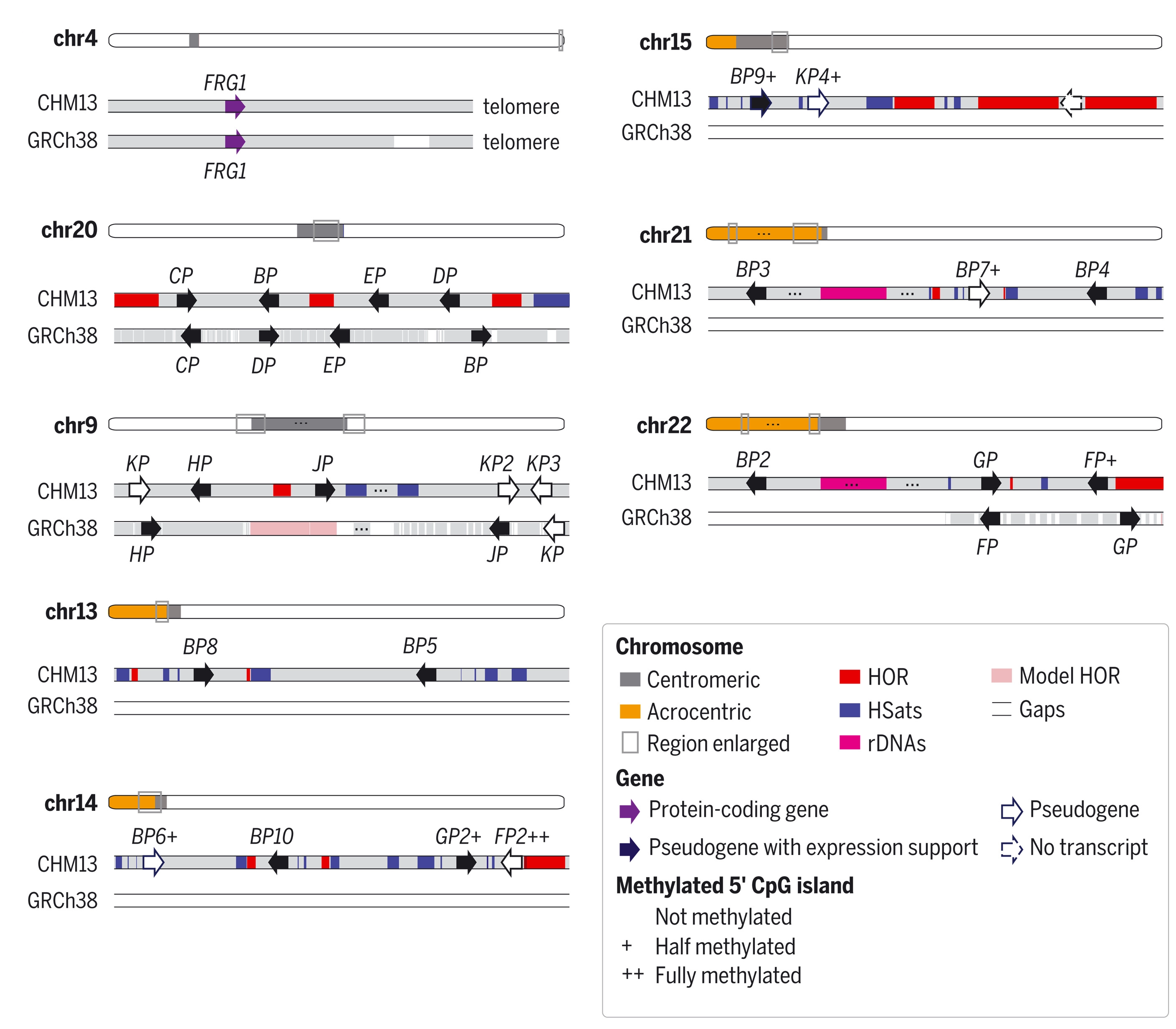
图:蛋白质编码基因 FRG1 及其在 CHM13 中的 23 个旁系同源物。之前的组装(GRCh38)中只找到了 9 个。基因绘制得比实际大小大,为简洁起见省略了 "FRG1" 前缀。所有旁系同源物都位于卫星阵列附近。FRG1 与面肩肱型肌营养不良症(FSHD)有关。图片来源:修改自 CC BY 4.0 via PMC [@t2t_human_genome_2022]
知识框 5.3:表型变异
小的(和大的)变异可以产生巨大的表型效应。

图:胡萝卜的颜色变异。图片来源:CC0 1.0 [@carrots_2006]
它们解释了我们周围生物体中可观察的变异。在农业中,变异被主动选择以创造我们今天在食物中看到的多种形状、颜色和口感品种。历史上,变异纯粹基于可见的表型进行选择,新的组合主要通过大规模杂交中的随机方式产生。
如今,通过基因组测序和变异检测获得的遗传信息越来越多地应用于植物和动物育种与选择。
变异¶
当(密切相关的)参考基因组已经可用时,读段可以映射到该基因组。映射涉及在基因组中找到与每个读段匹配的位置,允许一些小的差异——基因组变异。这种变异可以帮助解释表型变异(见知识框 5.3)。样品、个体和/或物种之间的基因组变异也可用于研究进化历史(另见关于多序列比对和系统发育的第 2 章和第 3 章)。变异分为两大类:结构性或大规模变异和小规模变异。首先,我们将重点讨论小规模变异。在这一组中,我们区分单核苷酸多态性(SNP)、多核苷酸多态性(MNP)和小插入缺失(indel):
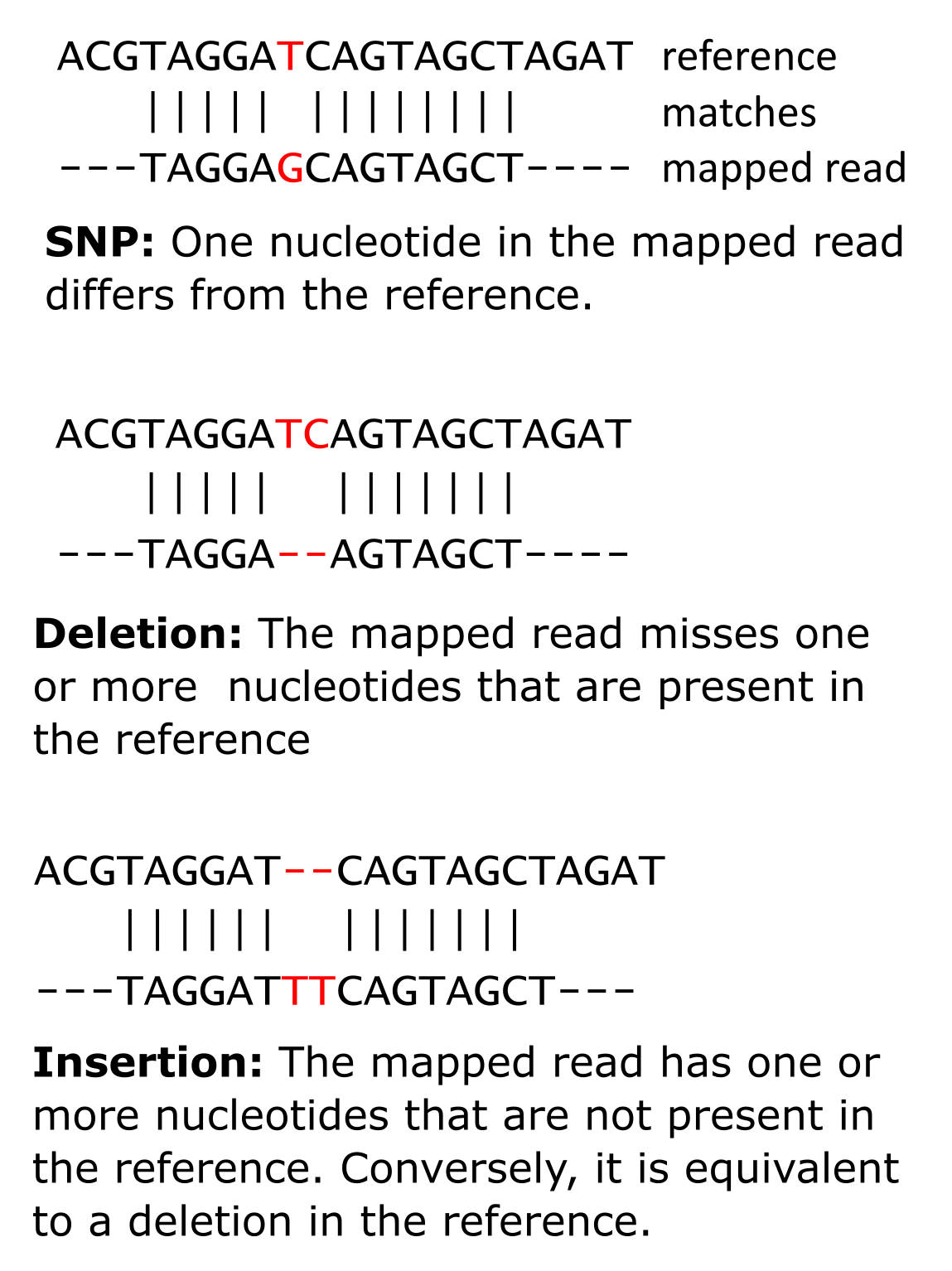
图:单核苷酸多态性(左上)、插入(下方)和缺失(右上)。图片来源:CC BY-NC 4.0 [@own_5_2024]
参考基因组特定位置的每个变异称为一个等位基因。在我们上面 SNP 的例子中(见图),我们有一个参考等位基因 T 和一个替代等位基因 G。关于 SNP,当样品来自单个个体时,参考基因组任何位置的理论等位基因数不能超过该个体的倍性:二倍体生物最多可以有两个不同的等位基因(如我们的例子),四倍体最多可以有四个,以此类推。鉴于我们只有四种不同的核苷酸,单个位置的可能等位基因的最大数量是四个,对于更高倍性来说等位基因会变得复杂。但如果我们发现二倍体生物中有超过两个等位基因,那一定是错误的结果,要么在读段中,要么在参考中。在极少数情况下,它可能是细胞间异质性的结果,例如癌症。
变异检测¶
正如我们已经看到的,变异可以是真实的,可以是测序错误的结果,也可以代表参考序列中的错误。检测变异(SNP 和 indel)并确定哪些是真实的或最可能是错误的过程称为变异检测。为了指示变异是真实的概率,每个变异被赋予一个质量分数。这考虑了假定位点的覆盖度和比对质量(即读段与参考的匹配程度)。变异检测软件还会报告一系列统计信息中的所谓等位基因频率(AF),由代表每个等位基因的读段占所有比对读段的比例决定。在具有一个参考等位基因和一个替代等位基因的二倍体生物中,我们期望两者平均频率为 0.5。当某个等位基因的频率接近 0 时,表明该变异很可能是由错误引起的。
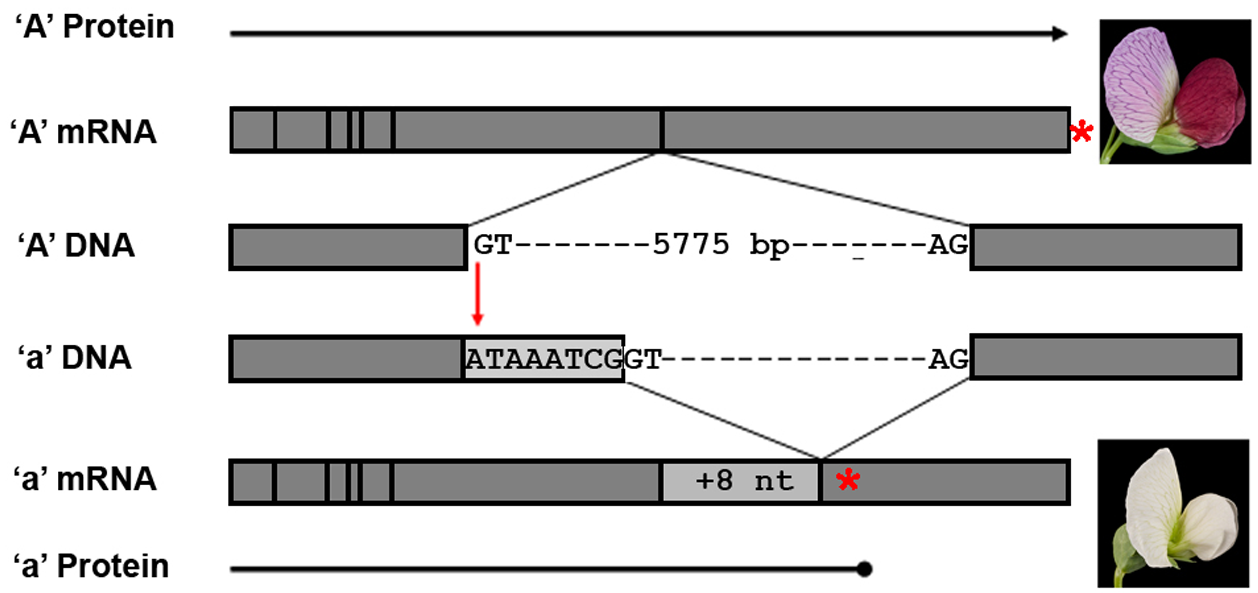
图:bHLH 基因的主要特征及其表达产物(不按比例)。在 Caméor 中,一种基因型为 a 的白花豌豆品种中,内含子 6 剪接供体位点有一个 G 到 A 的单突变,破坏了正常内含子加工所需的 GT 序列。在 DNA 中,外显子 6 和 7 显示为灰色方框,围绕内含子 6 剪接供体和受体序列。在 RNA 中,垂直线表示外显子连接处,浅灰色方框表示 a mRNA 中因内含子 6 错误剪接导致的 8 个核苷酸(nt)插入。红星表示预测蛋白质中终止密码子的位置,突出了白花品种中的提前终止。图片来源:CC0 1.0 [@flower_color_2010]
变异及其效应¶
个体之间的 SNP 是大多数表型变异的基础。有时一个变异就导致不同的表型,如经典孟德尔性状的花色(见上图)。更多时候,表型性状是多个变异的结果,一个例子是身高(在人类中,身高由超过 12,000 个 SNP 决定 [@yengo_saturated_2022])。一些变异可导致遗传缺陷或增加某些疾病的风险。研究充分的例子是 BRCA1 和 BRCA2 基因的突变(见下图)。BRCA1 基因中的特定突变使该人在一生中患乳腺癌的几率增加到 80%。
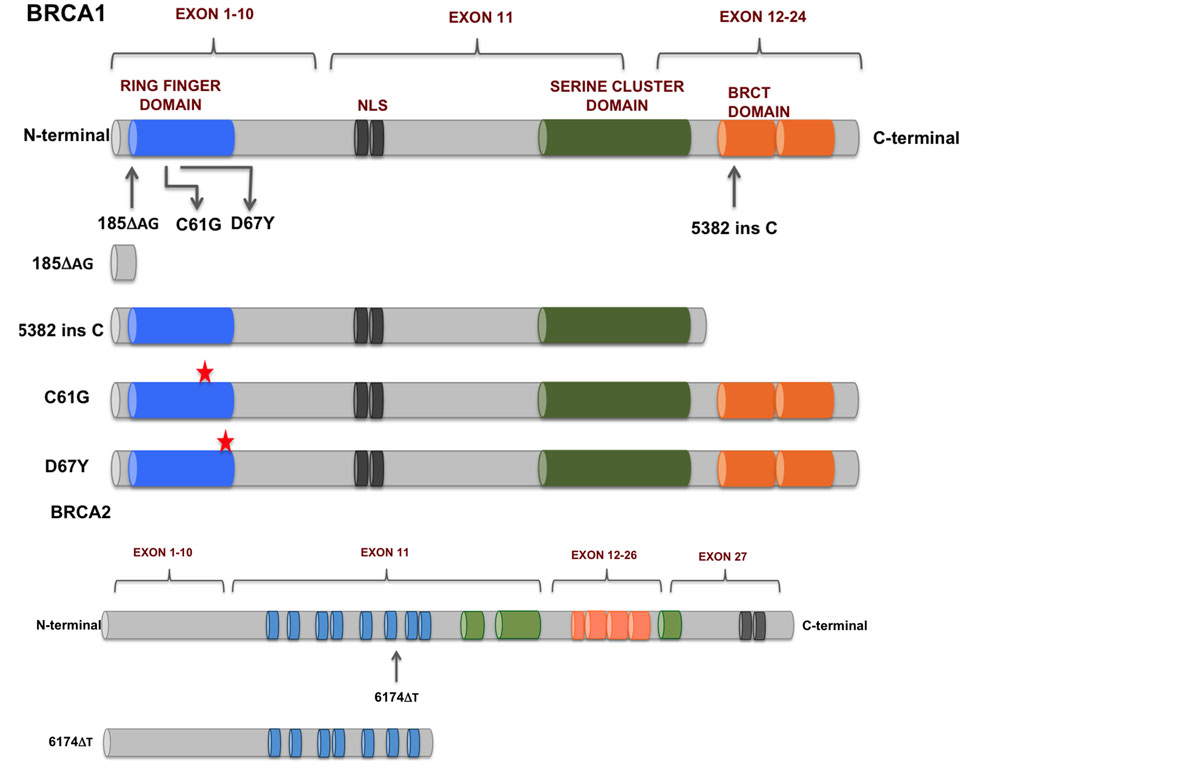
图:乳腺癌和卵巢癌中发现的 BRCA1 和 BRCA2 突变。图片来源:CC BY 4.0 [@BRCA_alt_2018]
大规模基因组变异¶
上面我们讨论了小规模变异,如 SNP 和 indel。相比之下,大规模变异是结构变异,其中基因组部分被重排、重复或缺失(见下图)。一种特殊情况是拷贝数变异,其中基因(或外显子)被重复。
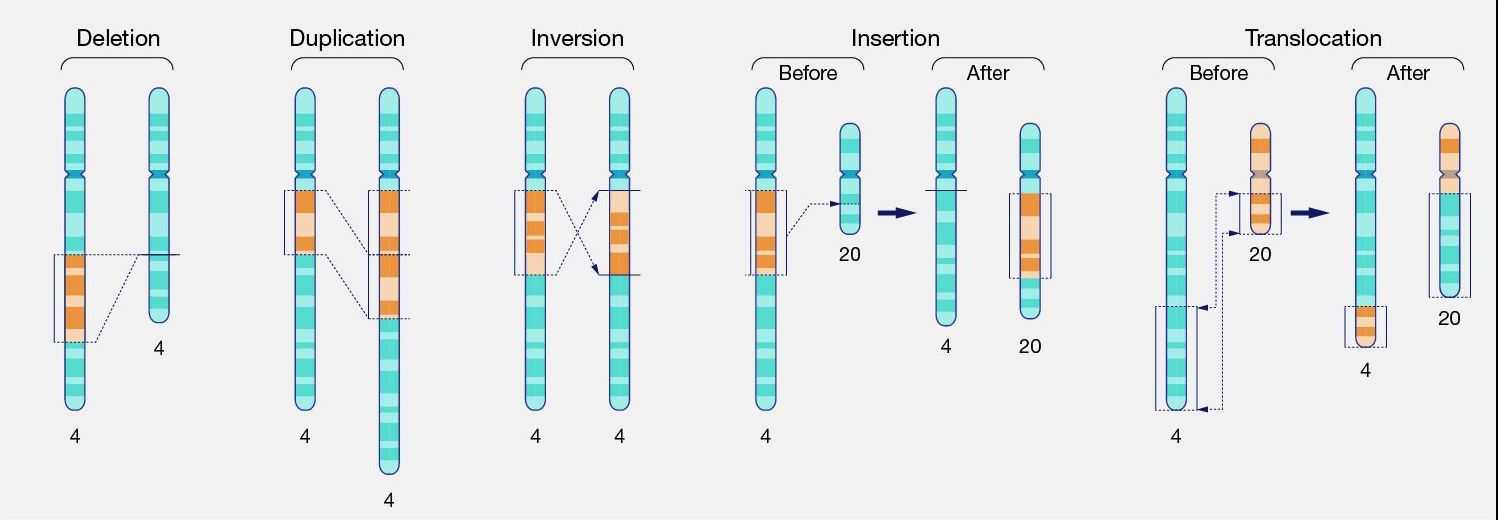
图:染色体水平不同类型的结构变异。图片来源:CC0 1.0 [@large_scale_variants_2024]
知识框 5.4:猫叫综合征
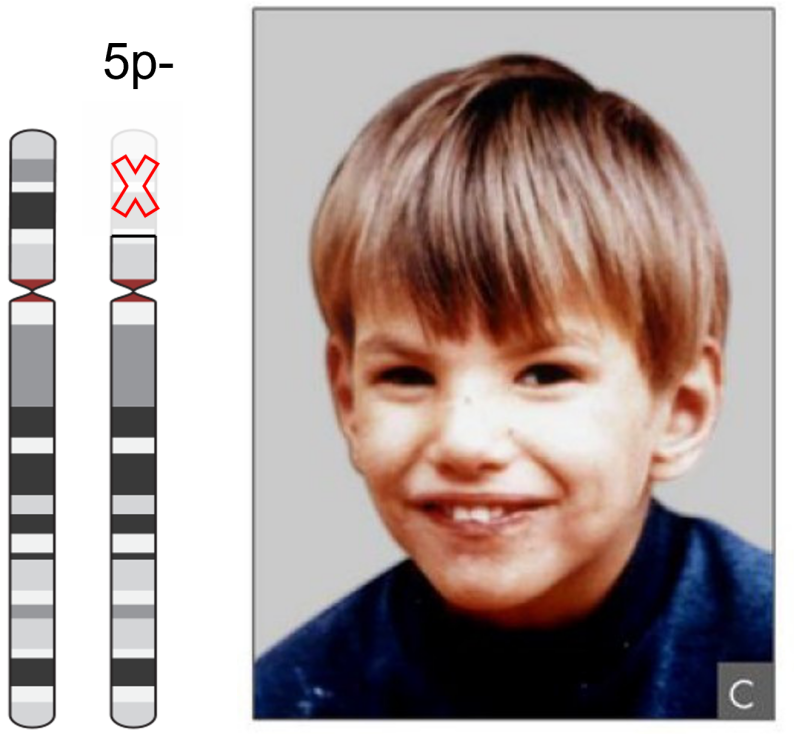
猫叫综合征是一种遗传性疾病,由 5 号染色体短臂的部分缺失引起。患有该病的婴儿发出高音调的哭声,听起来类似猫叫,因此得名该综合征。
此外,他们还受到生长迟缓和反射不良等困扰。图片来源:修改自 CC-BY 2.0 [@cri-du-chat] 和 CC-BY SA 4.0 [@chrom5]
结构变异¶
结构变异是大小超过约 1kb 的变异,可发生在染色体内和染色体间。结构变异有潜力对表型产生重大影响,例如疾病,但并不一定如此。结构变异似乎在癌细胞的发展中起着重要作用。
拷贝数变异¶
拷贝数变异(CNV)是结构变异的一种特殊情况,其中基因组上基因出现的次数发生变化。在二倍体生物中,缺失只留下一个拷贝,而重复可以导致个体中基因有三个或更多拷贝。改变拷贝数可以是群体中正常变异的一部分,例如参与免疫反应的基因的拷贝数存在变异;但更多时候,拷贝数变异会导致严重的表型变化,如人类疾病。
结构变异的检测¶
准确检测基因组中的结构变异并不容易。挑战在于检测变异的边缘(所谓的断点),以及在重复/插入/缺失的情况下,检测由此产生的拷贝数。当使用基于比对的方法时(在有参考基因组的情况下可行),我们可以利用读段深度和双端读段来检测变异。样品中基因拷贝越多,来自该基因的读段就越多(见下图),超过单拷贝的预期。反之,当一个基因的拷贝丢失时,覆盖度预期会下降。
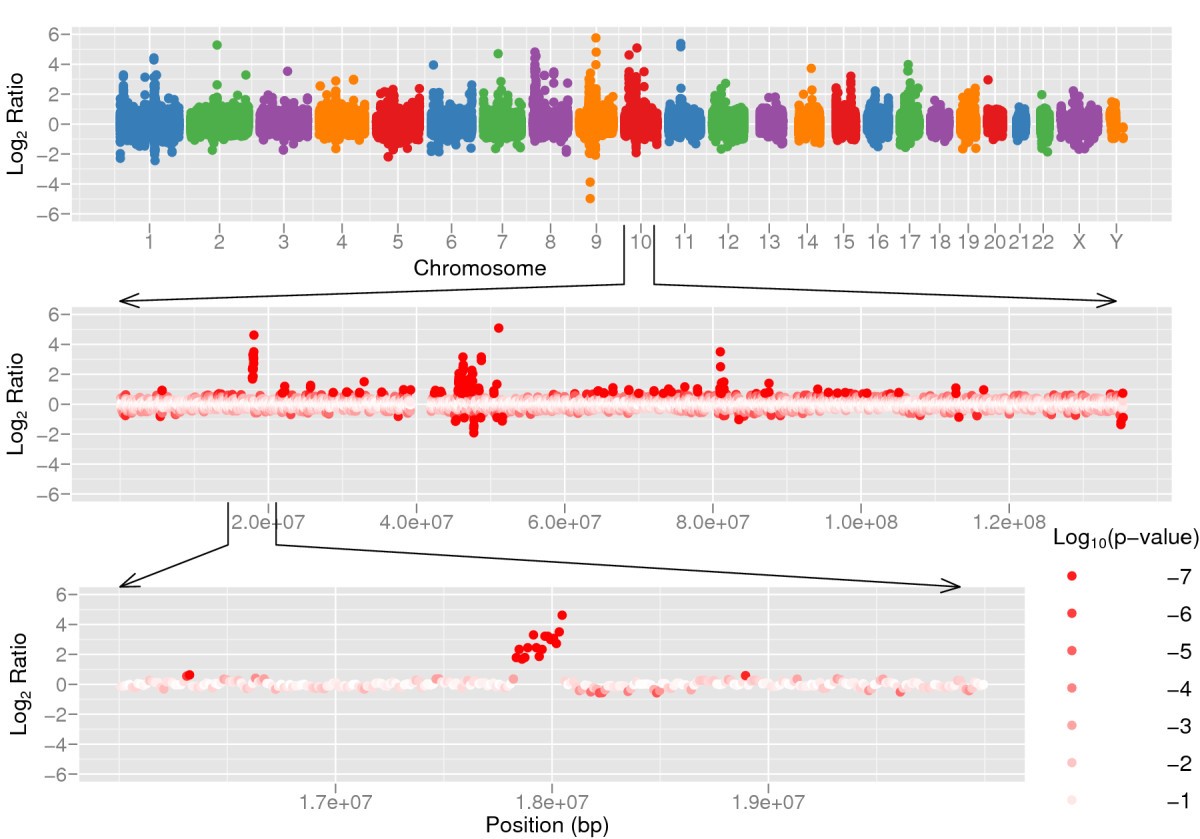
图:基因重复导致重复区域的覆盖度高于预期。图片来源:CC BY 2.0 [@gene_duplication_2009]
双端读段的方向以及分裂读段(读段的一部分比对到倒位区域,另一部分比对到周围序列)是检测倒位边界以及替换和易位的好指标。重排将导致一对读段中一条比对到一个基因组位置,另一条比对到另一个位置。跨越断点的读段在比对中会被分裂。
结构变异与拷贝数变异的示例¶
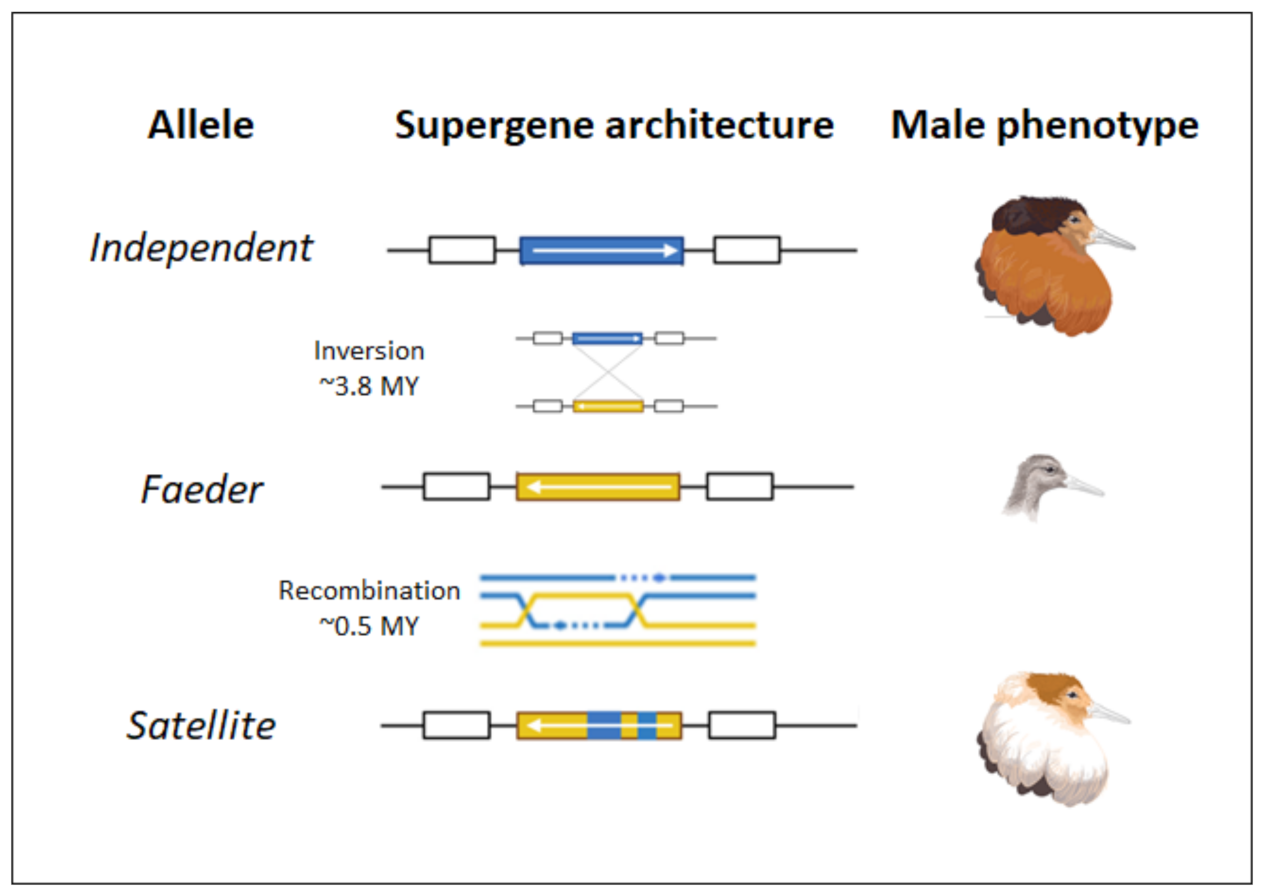
图:三种雄性形态中倒位的方向。图片来源:CC BY 4.0 [@male_morphs_2022]
大型染色体倒位在物种内表型变异中发挥作用,也被发现是两个不同物种杂交后基因渗入的结果。一个物种内倒位导致巨大表型差异的例子是瓣蹼鹬 (Calidris pugnax) 的三种雄性形态(见上图)。
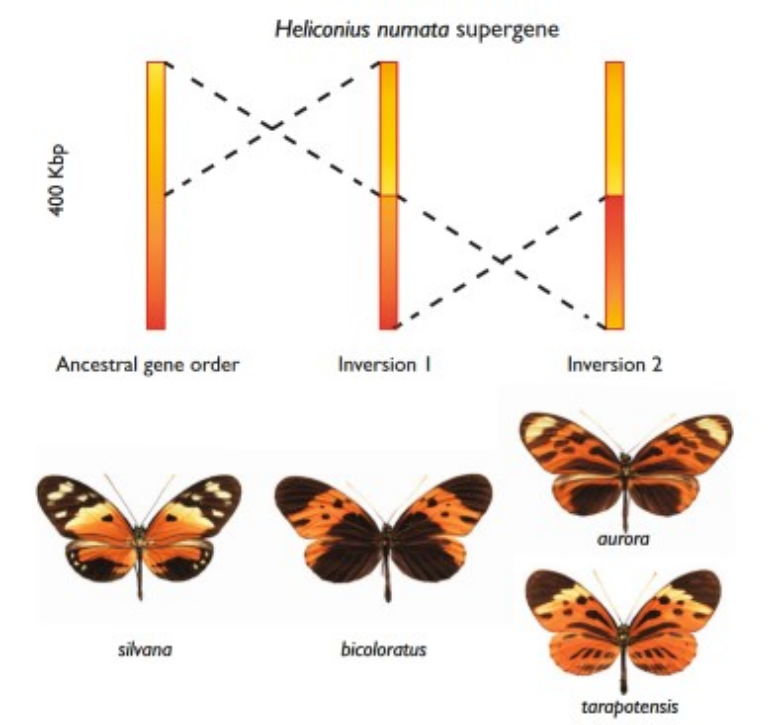
图:至少两个遗传倒位与 Heliconius numata 超基因相关。祖先基因顺序(与 H. melpomene 和 H. erato 中的匹配)显示在左侧,与祖先表型(如在 H. n. silvana 中发现的)相关。两个依次衍生的倒位与显性等位基因相关,显示在中间和右侧。图片来源:CC BY 4.0 [@butterflies_2017]
另一个例子是不同种类的 Heliconius 蝴蝶获得了包含翅色图案基因的倒位(见上图)。拷贝数变异也可以影响表型性状,例如小麦的开花时间、光周期敏感性和植株高度(见下图)。
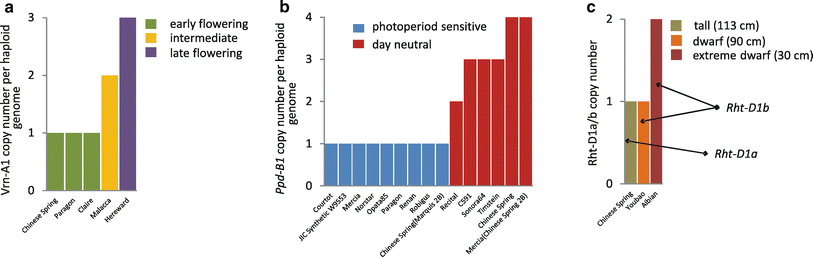
图:基因拷贝数变异(CNV)对小麦表型多样性的贡献。a) Vrn-A1 基因的 CNV 通过影响春化需求控制开花时间;b) Ppd-B1 的 CNV 通过影响光周期敏感性控制开花时间;c) Rht-D1b 基因(Rht-D1a 的截短版本)的 CNV 决定植株矮化表型的严重程度。在所有三种情况下,基因拷贝数对观察到的表型的影响已通过实验验证。数据来源:a, b, Díaz et al. (2012); c, Li et al. (2012)。图片来源:CC BY 4.0 [@CNV_2013]
功能基因组学与系统生物学¶
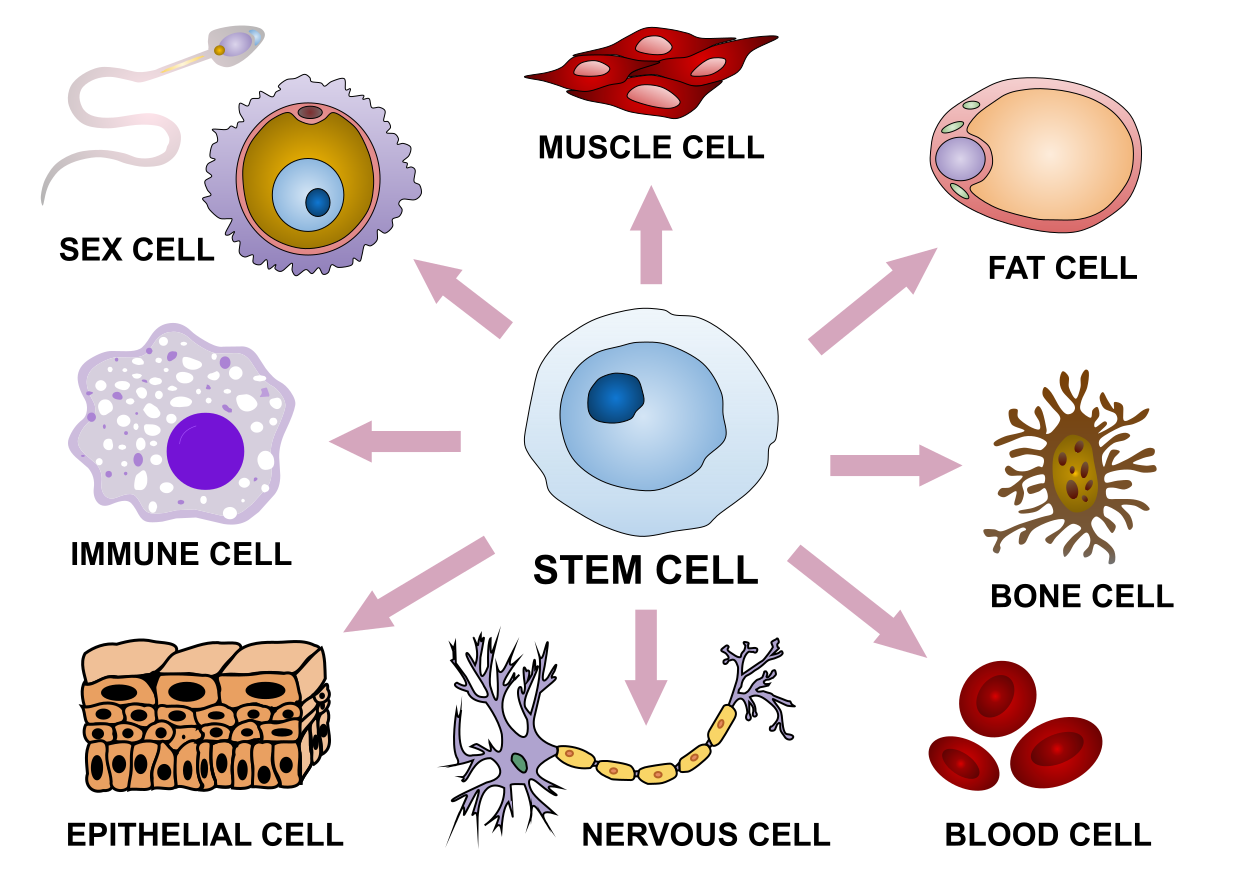
图:拥有相同的基因组,人类干细胞分化出各种不同的形态。图片来源:CC BY-SA 4.0 [@stem_cell_2019]
功能基因组学的必要性¶
虽然基因组学为我们提供了大量关于基因组的数据和基因信息,但这些显然只是故事的一部分。细胞不是静态对象:它们在生命周期中表现出不同的行为,并对环境变化和来自其他细胞的信号做出反应。在大多数多细胞生物体中,不同器官中的细胞以非常不同的方式发育,导致不同的细胞形态、组织组织和行为(见上图)。然而,每个细胞都包含相同的基因组,因此基因组的使用方式必然存在差异。换言之,如果基因组是生命的"书籍",那么它也必须包含如何阅读它的信息。
知识框 5.5:表观遗传学
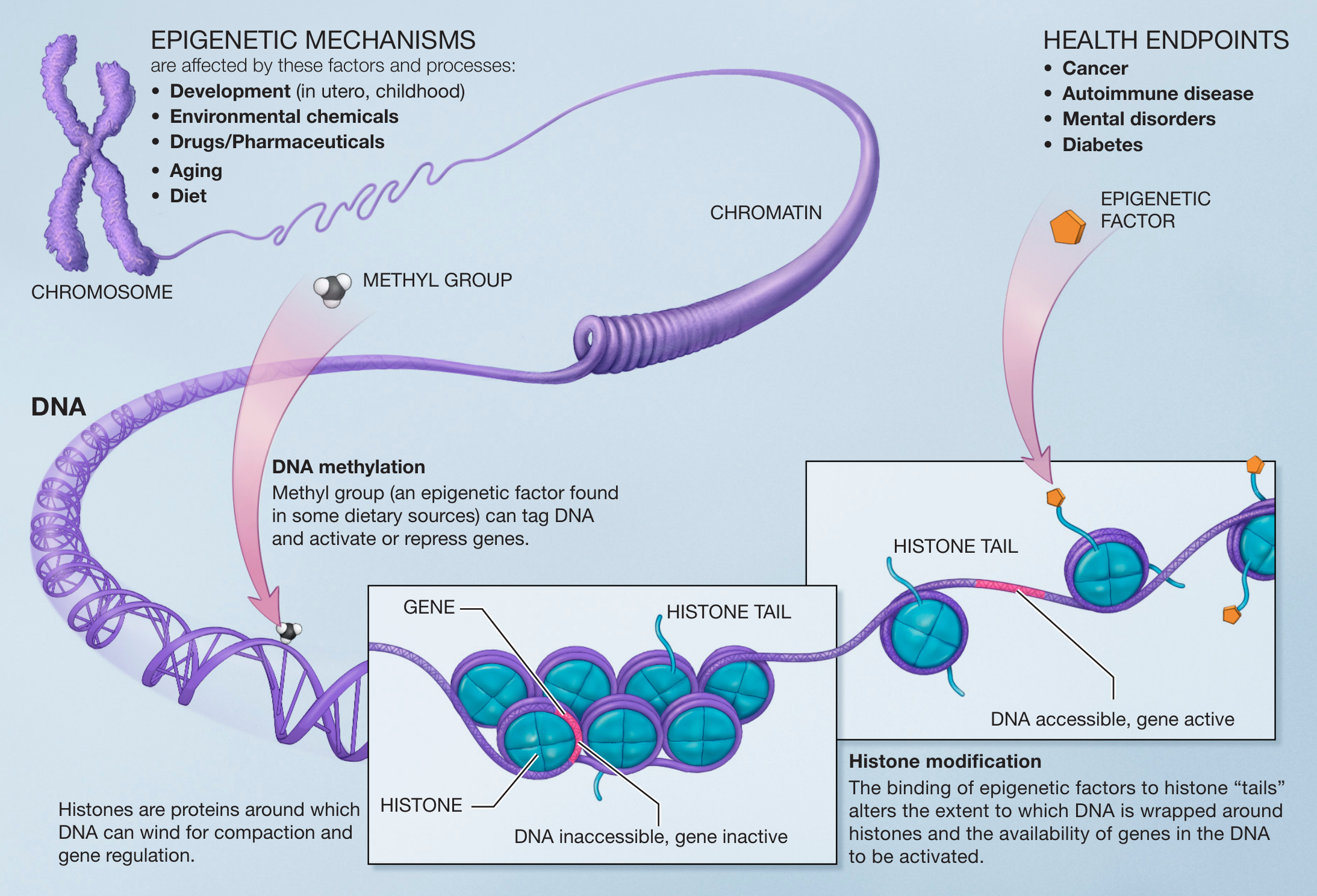
图:表观遗传机制受多种因素和过程影响,包括发育、环境化学物质、药物和药物、衰老和营养。DNA 甲基化是指甲基(在某些膳食来源中发现的表观遗传因子)可以标记 DNA 并激活或抑制基因。组蛋白是 DNA 可以缠绕其上的蛋白质,用于压缩和基因调控。组蛋白修饰是指表观遗传因子与组蛋白"尾部"的结合改变了 DNA 缠绕组蛋白的程度以及 DNA 中基因被激活的可用性。在更粗的层面上,DNA 的三维组织("染色质结构")也影响基因组的哪些区域可以被转录。在人类中,所有这些因素和过程都可以影响健康,其紊乱可能导致癌症、自身免疫性疾病、精神疾病或糖尿病等疾病。图片来源:CC0 1.0 [@epigenetics_2005]
部分解释在于所谓的表观遗传学——不改变 DNA 序列但影响基因表达的基因组修饰(见知识框 5.5)。除了表观遗传学之外,还有其他机制控制基因如何表达,以及产生的蛋白质最终如何在细胞中执行其功能。最知名的是蛋白质与 DNA 之间的相互作用(转录因子和增强子,影响表达);蛋白质之间的相互作用,形成复合物或传递信号;以及酶对代谢反应的催化。
研究基因如何被使用的领域称为功能基因组学。最突出的功能基因组学项目紧随人类基因组完成之后启动:ENCODE("DNA 元件百科全书")项目(2005-2015),旨在识别基因组的所有功能部分。
组学数据的作用¶
功能基因组学研究主要测量基因、蛋白质和代谢物的丰度及其相互作用。当在细胞范围的水平进行——即试图一次性测量所有某一类型的分子——这些称为组学测量。测量此类组学数据的技术通常是高通量的,这意味着很少需要人工操作或重复实验。我们通常区分五个主要的组学测量层级,如下图所示,尽管许多新的组学术语仍在不断涌现。除基因组学外,以下组学测量包括:
- 表观基因组学:基因组的所有表观遗传修饰。
- 转录组学:所有基因的表达水平。
- 蛋白质组学:所有蛋白质的存在/数量。
- 代谢组学:所有代谢物的存在/数量。
- 表型组学:细胞或生物体的最终表型(即形态或行为)。
此类测量越来越多地应用于混合样品,主要是细菌/真菌/病毒群落,如人体肠道和土壤中发现的。作为一种"元"分析,这被称为宏基因组学、宏转录组学等。
过去 25 年组学技术的引入拓宽了生物信息学领域,使其与所有生物学领域越来越相关。非常大的测量数据集现在被常规产生,需要进行清理、质量检查和处理。此外,数据应存储在数据库中,以便后续研究能够访问和重复使用。最后,对数据的仔细分析、解释和可视化对于帮助生物学家推断生物学功能至关重要。生物信息学为所有这些步骤提供了工具和数据库。
尽管组学测量提供了细胞状态和对扰动反应的高度详细的概览,但仍有若干重要局限:
- 实验成本:组学设备通常获取成本高,每次实验需要人力和耗材
- 技术噪声:所有测量技术都有固有变异
- 生物变异:不同的细胞、器官或个体在其生物状态和组成上会有差异
- 偏差和覆盖度:大多数组学技术对测量特定类型的分子或相互作用最有效(甚至只能用于此)
通常,功能基因组学实验涉及研究遗传变异对某些组学层面的影响。这种变异可以是天然的,例如比较两个已知因进化而有(有限)遗传差异的生物体的组学数据。也可以是实验引入的,例如在 DNA 序列中引入小突变、敲除基因、引入新基因等。变异也可以在环境中引入,例如改变温度、添加或去除营养物、引入药物等。在特定组学层面这些干预的效果提供了关于被操纵基因的功能或环境影响的信息。
从功能基因组学到系统生物学¶
功能基因组学利用组学测量来了解基因和蛋白质在细胞中的作用,但单个实验和测量通常只提供拼图的个别碎片,这些碎片在缺乏对其他细胞过程的理解时往往没有太多意义。认识到需要一种更加整体的方法,系统生物学被提出作为一种科学方法,其主要目标是构建活体系统的模型,通过假设形成、实验和模型扩展或修改来不断完善。
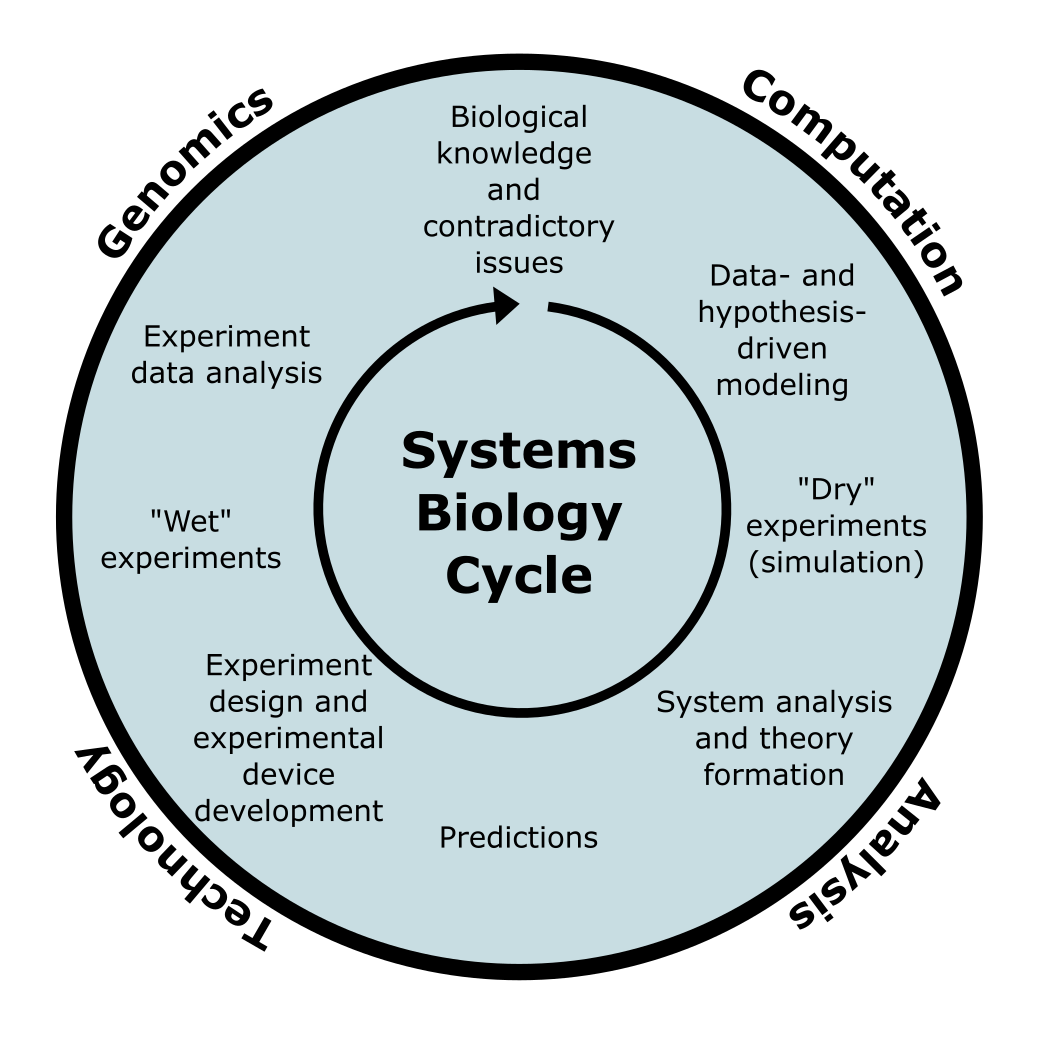
图:系统生物学循环,旨在迭代改进活体系统模型(基于 [@systems_biology_2002])。图片来源:CC BY-NC 4.0 [@own_5_2024]
最终,系统生物学的希望是达到对生命的系统层面理解,使我们能够模拟干预(突变、药物治疗等)的效果,甚至(重新)设计基因和蛋白质以改善某些行为,例如在生物技术中提高目标化合物的生产水平。虽然我们仍有很长的路要走,但组学数据分析是系统生物学中不可或缺的要素。
转录组学¶
转录组学关注测量转录本水平(即基因组转录为 RNA 的水平)。RNA 及其在细胞中的作用已在第 1 章中讨论。这里我们重点关注基因(即 mRNA)表达的测量和计数。理解转录组分析很重要的一点是,在真核生物中大多数基因含有内含子,且一个基因可以有多种替代转录本。
在转录组学中,目标是测量转录本的存在和丰度。这些测量基于大量细胞,但最近也可以研究单个细胞的转录组。那么转录本及其丰度能告诉我们关于研究对象的什么信息?在任何实验中,我们通常想知道细胞/组织/生物体在特定情况下的变化。最具信息量的是蛋白质水平,更具体地说是蛋白质活性,因为它们直接影响正在发生的事情。如下文将讨论的,检测和测量蛋白质很复杂。测量 mRNA 水平要容易得多,但重要的是要意识到它们只提供了细胞中发生的事情的代理指标。mRNA 水平并不总是与蛋白质水平相关,因为转录本可以被调控或抑制,影响翻译。蛋白质水平同样受调控影响,丰度并不总是与活性相关。
尽管有这些局限性,我们可以基于转录组学测量回答许多非常相关的问题。下面将讨论三种一般分析类型,但请注意,要检测实验条件与 mRNA 表达模式之间的关系,重要的是了解什么可能导致 mRNA 水平的变化。其中一些原因可能是有意的变异,有些则会产生噪声。
mRNA 水平:
- 是 mRNA 合成和 mRNA 降解的结果
- 在基因、亚型、细胞、细胞类型和组织以及发育阶段之间存在差异
- 随细胞周期、白天(昼夜节律)和/或季节变化
- 取决于环境
知识框 5.6:大型转录组学数据集
过去几十年中,若干重大国际科学计划已启动,旨在大规模测量基因表达(和其他组学层面)。希望我们能深入了解个体之间、组织之间以及疾病与健康样品之间的基因表达差异。鉴于医学研究的需要,这些工作大多在人类上进行;举几个例子:
- 癌症基因组图谱 (TCGA) 项目在 2006 年至 2018 年间运行。美国和加拿大的 20 个实验室的研究人员测量了 11,000 名患有 33 种常见类型癌症(包括 10 种罕见病)患者的基因表达,在正常和疾病组织中,产生了 2.5 Pb(拍字节)的数据。这导致了关于特定类型癌症新见解的多种发表,并最终形成了 Pan-Cancer Atlas。
- 癌症细胞系百科全书 (CCLE) 于 2008-2020 年运行,收集了约 1000 个用作人类癌症研究模型的细胞系的各种组学数据(包括转录组学)。这允许用户将 DNA 变异与转录本、蛋白质和代谢物的差异联系起来。
- 基因-组织表达 (GTEx) 项目始于 2013 年,旨在收集来自 54 种组织的多个捐赠者的基因组和转录组学数据。这允许研究人员深入研究 DNA 变异与组织特异性基因表达变异之间的关系。最新版本(V10)有来自近 1,000 个捐赠者的近 20,000 个样本。
在非医学研究中,也做了类似的工作。在植物科学中,例子包括 BAR——拟南芥的表达图谱,以及 1000 个植物转录组 (1KP) 项目。然而,非人类物种之间和内部的更广泛变异使得大规模生成有意义的数据更加困难。
如何测量 mRNA?¶
就像基因组研究一样,转录组学也从技术发展中受益匪浅,这些发展提高了测量的通量和灵敏度。如果你感兴趣,知识框 5.12 和 知识框 5.13 提供了对该领域发展重要的技术(如微阵列)的概述,但这些技术现在已不再广泛使用;目前,RNAseq 几乎专门用于测量 mRNA 水平。
注意微阵列现在已被 RNAseq 作为更便宜、更高质量的替代方案所取代(见下文)。然而,数据库中仍有许多微阵列样品可供重复使用,因为在发表科学论文时,将测量数据提交到此类数据库是强制性的。最知名的数据库是 NCBI 基因表达综合数据库 (GEO),截至 2024 年 3 月约有 710 万个样本,以及 EBI ArrayExpress。如果你对某个可能通过转录组学回答的问题感兴趣,首先应该查看已有的实验数据。注意所使用的技术决定了表达水平的解释方式(见下框)。
关于微阵列的重要知识
- 微阵列是表面上在特定位置附有特定 DNA 序列的器件,如果样品 DNA 链具有互补序列就可以与之结合
- 微阵列通过荧光间接测量表达;因此,测量可能有噪声且动态范围较低(即低表达水平无法被很好地测量)
- 某些微阵列类型比较两个样品并产生相对表达水平(倍数变化),通常进行 log2 转换,使 0 表示无变化,+1 表示 2 倍高表达,+2 表示 4 倍高表达,负数表示低表达
- 其他微阵列类型测量代表单个样品中绝对表达的值(以任意单位);在比较样品之间的测量时,标准化很重要
RNAseq¶
RNAseq 利用经济可靠的测序方法。对 RNAseq 发展重要的是 NGS 方案和测序仪的可靠定量特性。RNAseq 是无目标的:原则上样品中的所有 RNA 都可以被测序,不需要预先了解转录本序列。虽然 RNAseq 主要用于研究转录本丰度,它也可以用于检测转录本亚型(及其丰度)以及变异(见上面的变异)。
实验方案¶
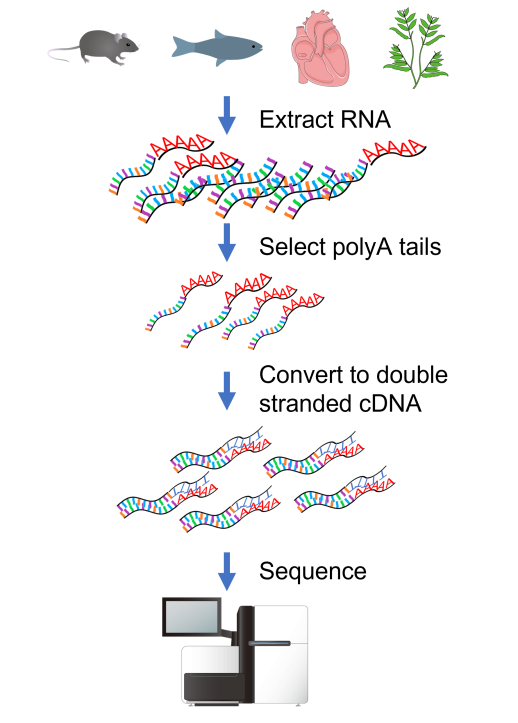
图:标准 RNAseq 方案。图片来源:CC BY 4.0 [@own_5_2024] 修改/创建自 CC BY 4.0 [@RNAseq_seq_2011, @RNAseq_plant_2023, @RNAseq_seq_2020], CC BY 3.0 [@RNAseq_mouse_2013, @RNAseq_heart_2016], CC0 1.0 [@RNAseq_fish_2014]
RNAseq 实验的标准方案如上图所示。首先,从生物样品中提取所有 RNA(总 RNA)。然后,使用 polyT 寡核苷酸选择具有 polyA 尾的 RNA 来富集 mRNA。RNA 随后被转换为稳定的双链 cDNA。所得的 cDNA 文库通常以 100-150bp 的双端读段进行测序。标准测序运行每个样品产生 3000 万条或更多读段。
目前使用的读段长度相对较短,需要复杂的方法将读段分配给外显子和亚型。该领域的新发展是长 cDNA 转换,允许在 PacBio 上测序全长转录本,以及在 Oxford Nanopore 上直接测序 RNA。这使得检测样品中实际存在的亚型成为可能。
接下来,读段需要被分配到对应的转录本。为此有两种选择:将读段比对到现有的参考序列(可以是基因组或转录组);或转录本的从头组装(类似于基因组的组装)。一旦读段被分配到对应的转录本或基因,表达量通过计算每个特征的读段数来定量。
不使用探针(与 qPCR 和微阵列相比)的优势是 RNAseq 可以在没有参考基因组的物种中使用,可以识别可变剪接转录本、转录本中的 SNP 等。挑战是通常产生大型数据集,需要专门的分析流程。
比对¶
原则上,来自 RNAseq 实验的测序读段与从基因组 DNA 测序的读段没有区别,它们可以被比对到参考序列。当将 RNAseq 读段比对到组装的转录组(只包含 RNA 序列的参考序列)或原核生物基因组时,适用相同的算法。
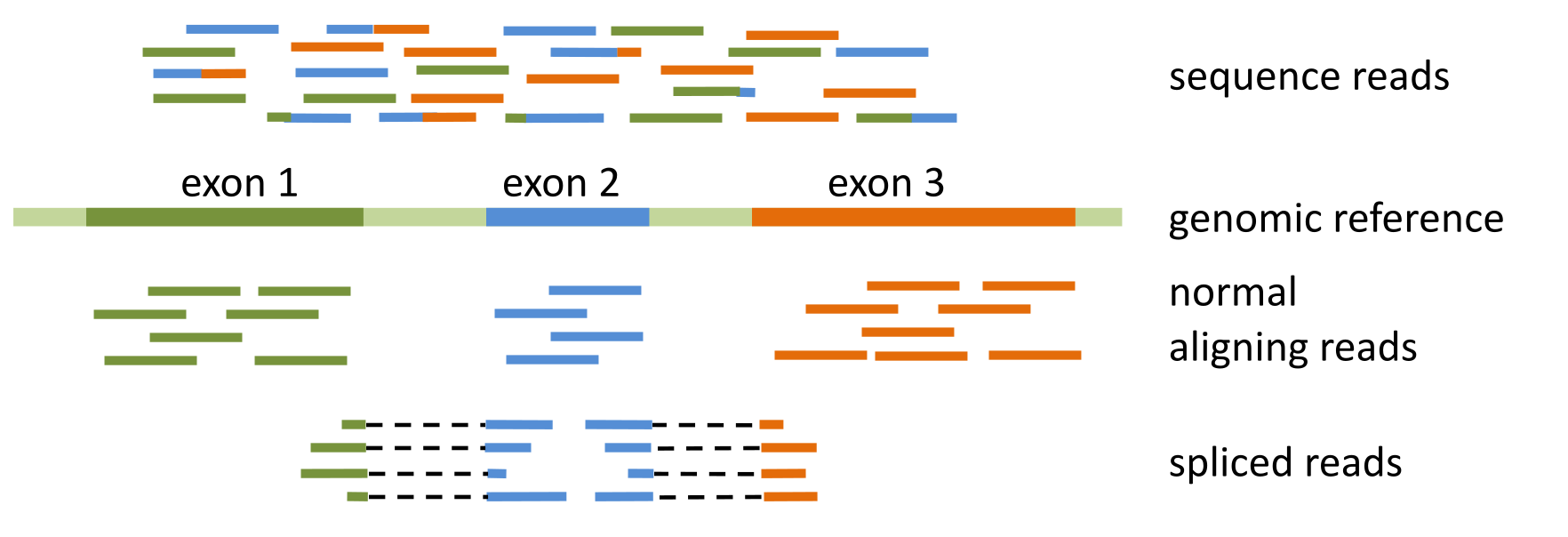
图:mRNA 读段到基因组参考的剪接比对,使用剪接感知比对器。图片来源:CC BY-NC 4.0 [@own_5_2024]
将真核生物 mRNA 序列比对到基因组参考更加复杂,因为大多数基因含有内含子,这些内含子在成熟 mRNA 中不再存在(见上图)。这意味着读段可能包含外显子-外显子连接处,应在参考上被分裂,即所谓的剪接比对。大多数比对器不会将此视为有效选项。出于这个原因,开发了专门的剪接感知比对器,能够比对正常地连续比对到参考序列的读段,也能比对跨越剪接位点被分裂的读段(见上图)。它们还考虑已知的内含子-外显子边界来确定读段需要在哪里分裂以及分裂比对是否正确。
转录本定量¶
测序和比对之后的下一步是定量转录本的丰度,即表达水平。分配到每个特征(外显子或基因)的读段被计数,其基本假设是比对到某个特征的读段数量与该特征在实验中的丰度强相关。在不同样品、条件和实验之间比较转录本丰度并不像看起来那么简单。除了影响 mRNA 丰度的上述因素外,每次测序实验中都存在变异。影响样品间读段计数可比性的主要变异是每个样品中测序的总读段数。此外,并非所有转录本长度相同,这影响了每个转录本检测到的读段数。因此,需要进行标准化以考虑这些差异并使数据具有可比性。主要方法包括:
-
简单计数:这是每项分析的起点。我们计算比对到每个外显子或基因的读段数。
-
CPM:每百万读段计数 (counts per million),一种相对度量,对读段计数根据样品的总读段数进行校正。它为每个读段分配一个值,对应于该读段占总读段数的比例。这个微小分数随后乘以一百万以便于阅读。
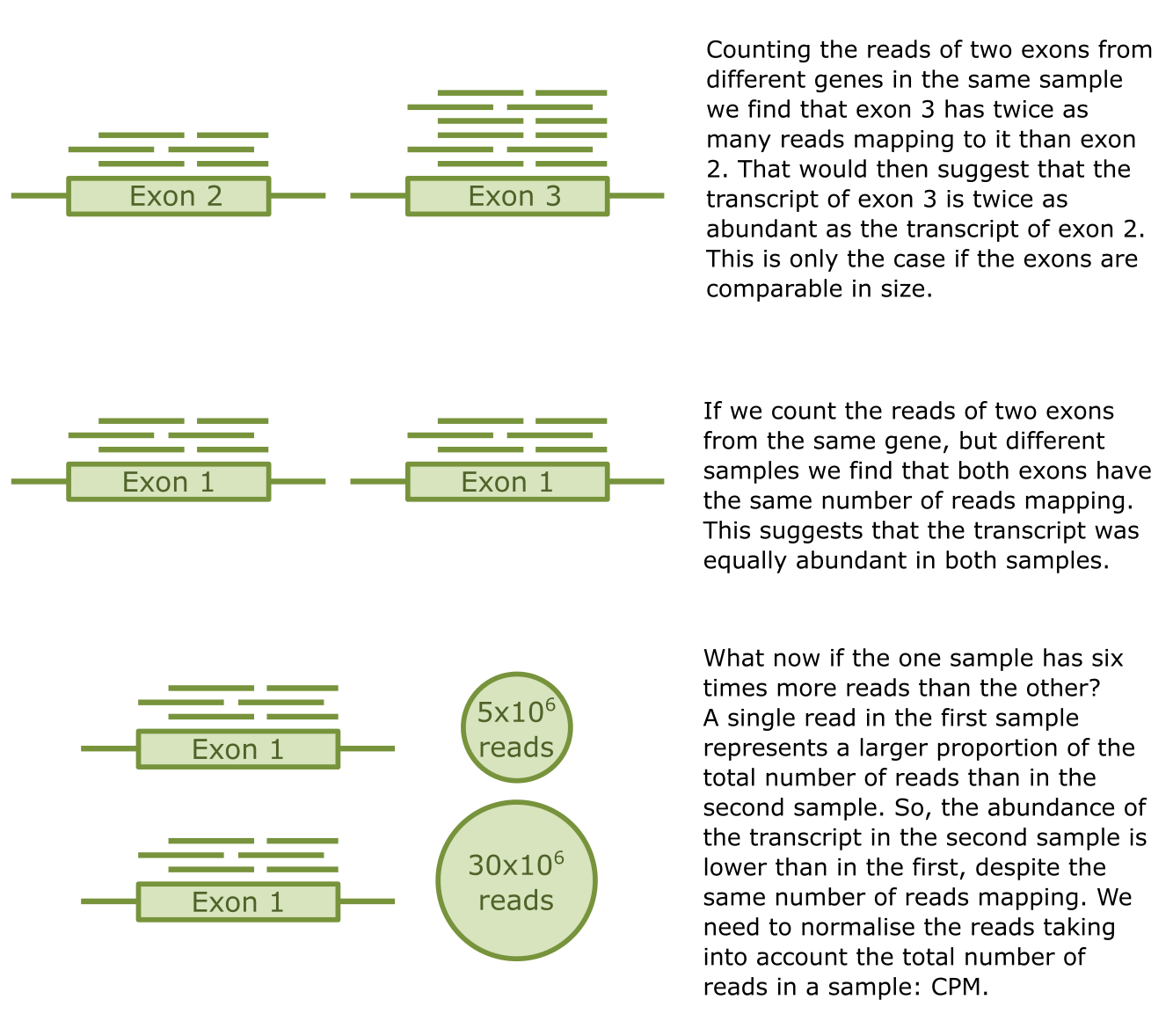
图:图片来源:CC BY-NC 4.0 [@own_5_2024]
- RPKM、FPKM 和 TPM:当比较两个不同转录本的表达时,我们还必须考虑所比较转录本的特征并进行相应的标准化。RPKM 和 FPKM(每千碱基转录本每百万读段/片段数)根据特征长度和总读段数进行标准化。TPM(每百万转录本数)按转录本进行标准化。TPM 使用一种计算方法,给出每个转录本占原始样品中总转录本数量的比例的度量。

图:图片来源:CC BY-NC 4.0 [@own_5_2024]
没有明确的最优方法,关于 RPKM/FPKM 和 TPM 哪个更优有很大的争论。CPM 显然只能在转录本长度没有差异时使用,例如在两个样品之间比较同一转录本时。
知识框 5.7:更多细节
到目前为止,大多数 RNAseq 实验都在细胞群上进行,因为测序设备需要大量的 DNA。这意味着不同细胞类型或不同生命阶段的细胞被包含在单个样品中。虽然用这种方法检测差异表达基因显然是可行的,但微弱或更细微的变异会在所有细胞中被平均化。最近,可以将组织分离成单个细胞并分别测序每个细胞的方法已成为可用。这确实需要通过 PCR 扩增 RNA 以达到测序所需的量,以及复杂的生物信息学和统计方法来处理由此产生的数据。其他新技术允许在特定位置(如组织中的网格点)空间性地测量转录。关于单细胞转录组学的综述,请参见这篇论文;关于空间转录组学的近期综述可在这里找到。
蛋白质组学¶
如前所述,转录组学被广泛应用但不能准确反映整体细胞状态。产生的蛋白质是细胞的主力军,了解它们的浓度、修饰和相互作用比基因表达能提供更多见解。遗憾的是,虽然蛋白质组学技术已取得重大进展,但准确测量蛋白质仍然复杂且昂贵,原因如下:
-
显然,识别具有 20 种构建单元(氨基酸)的分子比识别具有 4 种构建单元(核苷酸)的分子更困难。
-
此外,鉴于可变剪接,需要区分的蛋白质数量高于基因数量。
-
蛋白质可以在翻译后以多种方式被修饰——在结构上和生化上,通过在单个氨基酸上添加许多不同的基团。对于某些蛋白质,估计在细胞中可以找到数千种不同的变体。
-
大多数蛋白质是有结构的,有些形成复合物或嵌入膜中;这些结构通常需要被去除以允许准确测量。
-
蛋白质缺乏使 DNA 和 RNA 容易复制(PCR)和测量的特性:复制和与互补链的结合。
-
蛋白质丰度的动态范围巨大:某些蛋白质浓度比其他蛋白质高百万倍。如果我们有复制低丰度蛋白质的方案(如 DNA 的 PCR),这不会是问题,但这样的方案并不存在。
尽管如此,已有多种测量蛋白质及其相互作用的方法在使用。我们区分定量蛋白质组学(测量蛋白质的存在/缺失和水平)和功能蛋白质组学(测量蛋白质与其他分子的相互作用)。
定量蛋白质组学¶
虽然一些较早的基于凝胶的技术已被用于测量蛋白质的缺失/存在甚至水平,但这些已被质谱(MS)设备所取代。自 19 世纪末问世以来,这些设备一直在不断发展和改进,现在已达到允许研究各种大小分子的准确度水平,也包括代谢组学。它们在具体配置上有所不同,但都遵循三个基本步骤:
- 电离分子。
- 基于质量分离或选择分子。
- 检测到达检测器的时间和/或位置以推断每个分子的质量。
严格来说,MS 测量的是质荷比(m/z)而非实际质量,即质量与离子电荷数的比值。
每个步骤有不同技术可用,最适合检测特定的混合物、感兴趣的化合物(蛋白质、代谢物)和化合物大小范围。下图展示了几种广泛使用的分离步骤,即通过测量飞行时间或对磁场偏转的敏感性,或通过调节振荡电场以仅允许特定质量通过。
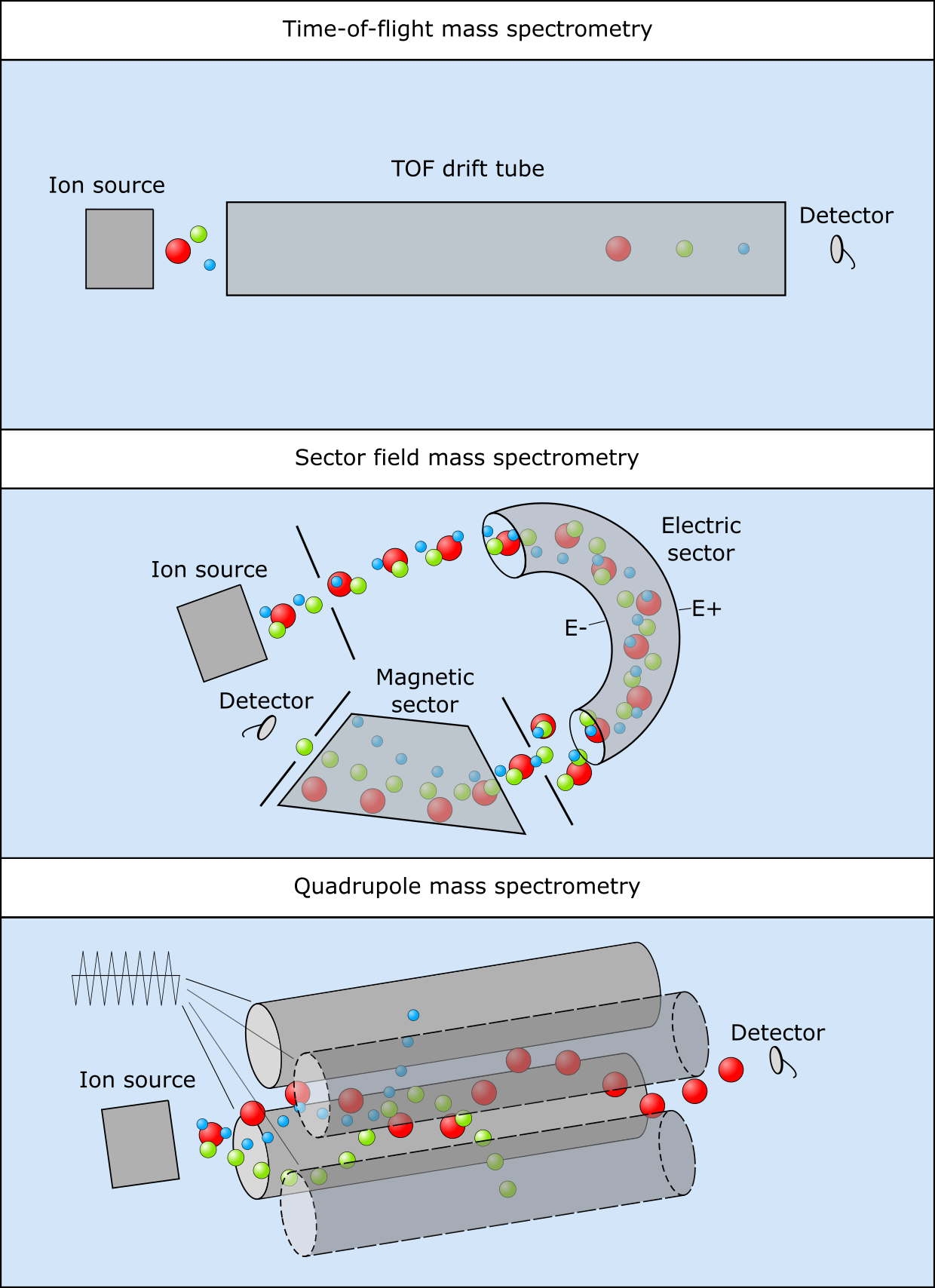
图:三种质谱设置,(上)飞行时间、(中)扇形场和(下)四极杆。图片来源:CC BY-NC 4.0 [@own_5_2024]
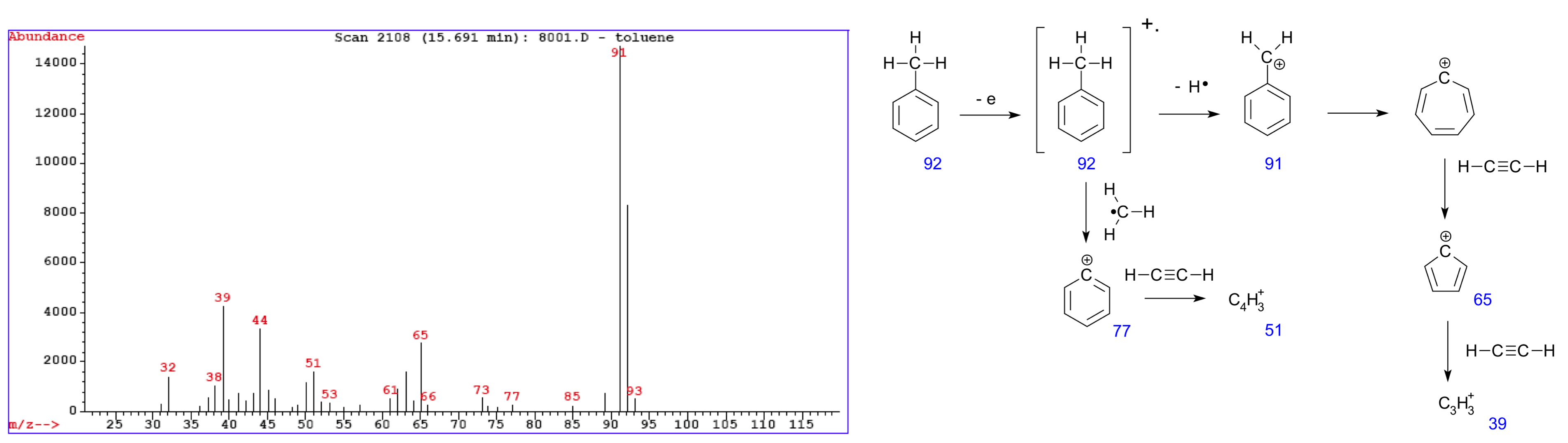
图:在甲苯上测量的质谱示例(左)。各个峰对应原始分子的碎片(右)。图片来源:修改自(左)CC BY-SA 3.0 [@mass_spectrum_left_2005],(右)CC BY-SA 3.0 [@mass_spectrum_right_2008]
任何 MS 实验的输出是质谱图,x 轴为 m/z 比值,峰表示检测到多少个特定质量的分子(见上图)。
理论上,如果有一个已知分子式(如蛋白质或肽段)的数据库及其计算质量的数据库,可以查找每个质量并识别相应的分子。解释此类谱图的一个主要挑战是 MS 设备的分辨率有限,这意味着某个峰仍可能由许多不同类型的分子引起。某些感兴趣的较小分子甚至可能具有相同的质量(如亚型),因此无法区分,这在复杂混合物中尤其困难。一些扩展方法试图解决这个问题;这些在知识框 5.14中有更详细的解释。
在生物信息学中特别感兴趣的是鸟枪法蛋白质组学,本质上类似于鸟枪法基因组学。特别是对于蛋白质,这是一种协议,首先使用酶在特定位点切割蛋白质(例如,胰蛋白酶在精氨酸和赖氨酸处将蛋白质切割为肽段)(见下图)。然后测量肽段质量,并与从已知蛋白质数据库预测的肽段质量谱进行比较,以识别可能正在测量的蛋白质。这种方法也可用于测量翻译后修饰,因为它们导致修饰肽段的测量谱中出现小的(已知的)位移。
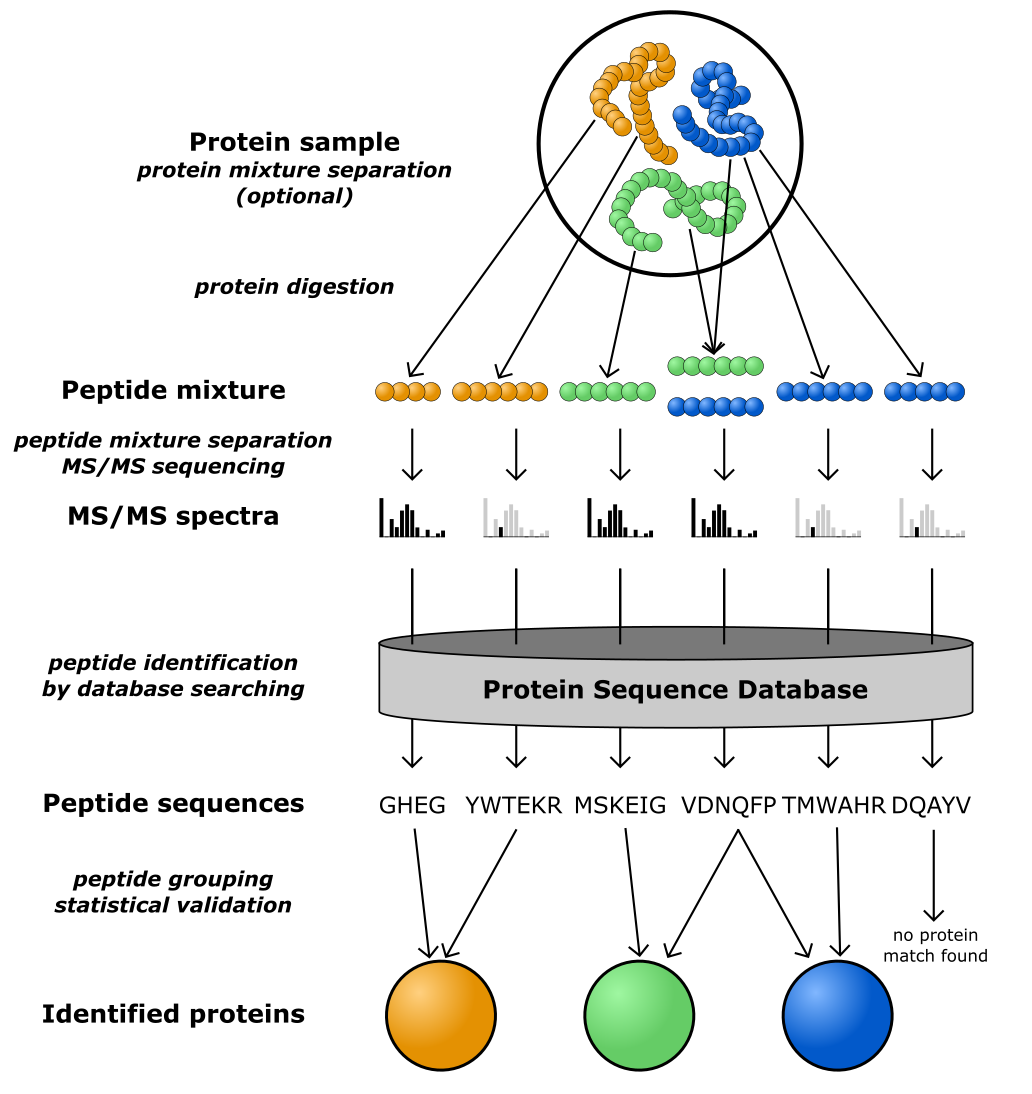
图:鸟枪法蛋白质组学的示意图。图片来源:CC BY-NC 4.0 [@own_5_2024]
功能蛋白质组学¶
除了蛋白质水平外,我们还对蛋白质在细胞中做什么感兴趣:它们的功能和相互作用。为此已开发了许多协议和分析方法,大多数集中在蛋白质-蛋白质、蛋白质-DNA 和蛋白质-代谢物(酶促)相互作用。知识框 5.15 列出了一些测量此类相互作用的方法。请注意,虽然许多这些实验很繁琐,但它们对推进功能基因组学至关重要——(生物信息学)预测严重依赖高质量数据,不能替代实验验证。
与转录组学数据一样,"互作组学"测量存储在数据库中,如 IntAct,可用于获得细胞范围蛋白质相互作用网络的见解(见下图)。高度连接的蛋白质组——即具有许多相互作用的——可以指示蛋白质复合物或细胞内或细胞间的信号通路;蛋白质-DNA 关系可用于识别基因表达调控程序。
请注意,上述方法测量的是蛋白质之间的物理相互作用,而非功能相互作用。当两个蛋白质具有相似功能时会发生功能相互作用——即使它们可能从未真正物理上相互作用,例如当它们是同一基因的两个替代转录因子时。这种功能相互作用可以在一定程度上测量,但主要由生物信息学工具预测,这些工具结合各种证据:文献、序列相似性、基因共表达等。STRING 和 GeneMania 是最知名的例子。

图:果蝇的蛋白质相互作用网络(4,927 个蛋白质,209,912 个通过串联亲和纯化发现的相互作用),颜色标示对应蛋白质复合物的簇。图片来源:修改自 [@protein_network_2011],使用 Elsevier 用户许可
代谢组学¶
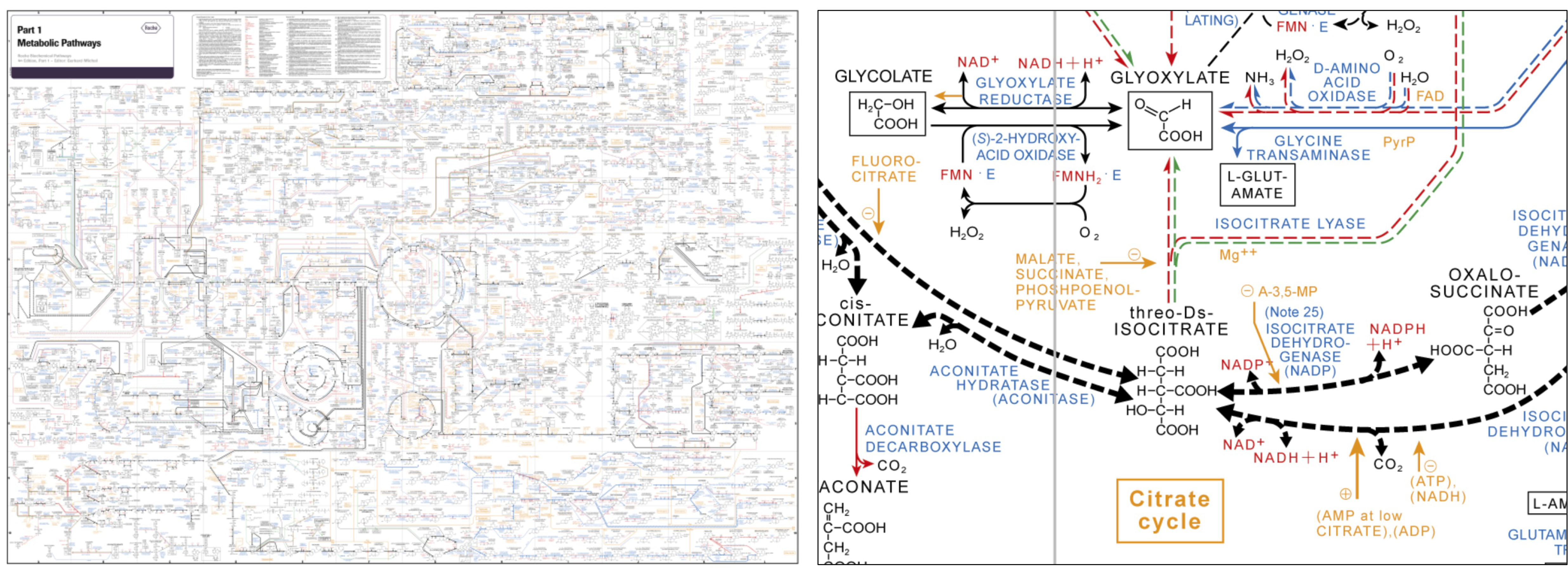
图:Roche 生化途径图表:代谢过程的全局概览(左)和柠檬酸循环一部分的特写(右)。图片来源:[@metabolic_network_2016]
许多细胞产生多种代谢物——参与代谢的小分子或化合物。许多所谓的初级代谢物作为基本分子(如 DNA 或蛋白质)的构建单元,并为反应提供能量。其他代谢物——特化代谢物——在许多生物体中用于通讯、调控(激素)、防御(抗生素)和共生。一些代谢物还调控相关表型。因此,解析细胞中循环的所有分子的结构并测量代谢物的浓度——作为细胞组织的"终点"——对于研究生物体和群落的生长和发育高度相关。代谢组学在医学和药理学、食品安全以及揭示工业生物技术中微生物的生产谱方面也很重要。
对于代谢物的测量,主要采用上述质谱技术(见上文),特别是 GC-MS 和 LC-MS(见知识框 5.14)。由于代谢物大小和特征的范围很大,且许多代谢物仍然未知,从质谱中鉴定它们仍然非常具有挑战性。一个优势是已知的代谢反应——收集在代谢网络中(见上图)——可以支持系统生物学方法,特别是在微生物中。
表型组学¶
细胞调控的最终结果是表型,即细胞或生物体在宏观尺度上可观察的特征或行为的集合。这些表型通常以复杂的方式依赖于细胞中大量分子的水平和相互作用。揭示基因型-表型关系——即基因组层面的什么变异导致了(被破坏的)表型——是许多科学领域(包括医学)最重要的目标之一。
不同生物体的潜在表型集合是巨大的,并且没有像其他组学层面那样的标准化表型组学方法。例外包括人类疾病和遗传障碍的结构化数据库,如 MalaCards 和 OMIM。类似的方法开始进入生物学的其他领域(生态学、植物发育和育种以及动物行为),包括图像、视频和跟踪数据的(标准化)存储库。可靠的高通量表型组学数据将被证明对于理解我们发现的遗传变异至关重要。
组学数据分析¶
转录组学、蛋白质组学和代谢组学(可以)都提供关于分子水平的定量测量。所得数据可以以各种方式分析,以回答不同的问题。主要方法包括:
- 可视化:便于检查实验结果和识别大的模式。
- 差异丰度分析:比较条件、细胞类型或菌株之间的丰度水平。
- 时间序列分析:跟踪随时间的变化(即时间序列实验)。
- 聚类:基于丰度的相似性对基因或样品进行分组(例如,以了解共享功能)。
- 分类:找出哪些基因可以预测特定表型(例如,疾病)。
- 富集分析:了解在给定基因集合中哪些生物学功能/过程最常出现。
下面我们将逐一讨论。大多数示例将以转录组学数据提供,我们将始终使用"基因"一词,但这些方法可以不加修改地应用于定量蛋白质组学("蛋白质")和代谢组学("代谢物")数据。
但是,请记住所有这些测量都是有噪声的,应小心区分感兴趣的生物变异与不相关的技术和生物变异。作为示例,RNAseq 通常作为实验的一部分进行,目的是找到对两种或多种实验条件有不同响应的基因。此类实验旨在排除尽可能多的变异,但仍然会检测到一些不是处理结果而是测量噪声的丰度差异。为了清楚区分真实差异和噪声,相同条件的重复测量很重要(见下图)。这些被称为重复样品。重复测量变异的基础是生物和技术变异;下图对样品中检测到的所有转录本进行了比较,两个重复样品(左)和两个不同条件(中和右)。
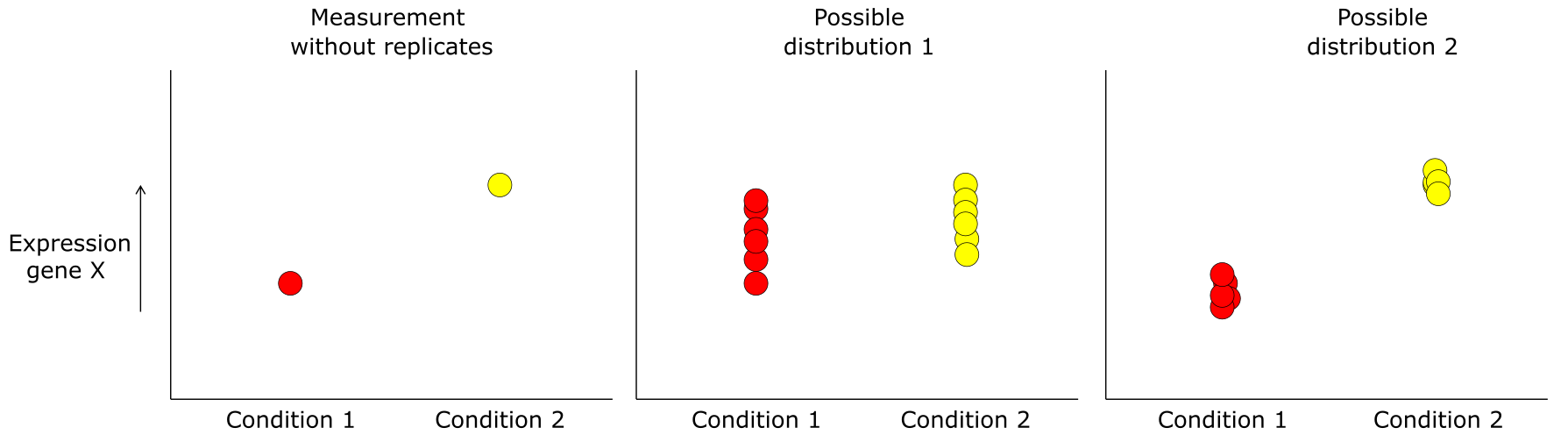
图:无重复测量时基因 X 在两个条件间表达差异,以及该测量可能来自的两种可能分布。图片来源:CC BY-NC 4.0 [@own_5_2024]
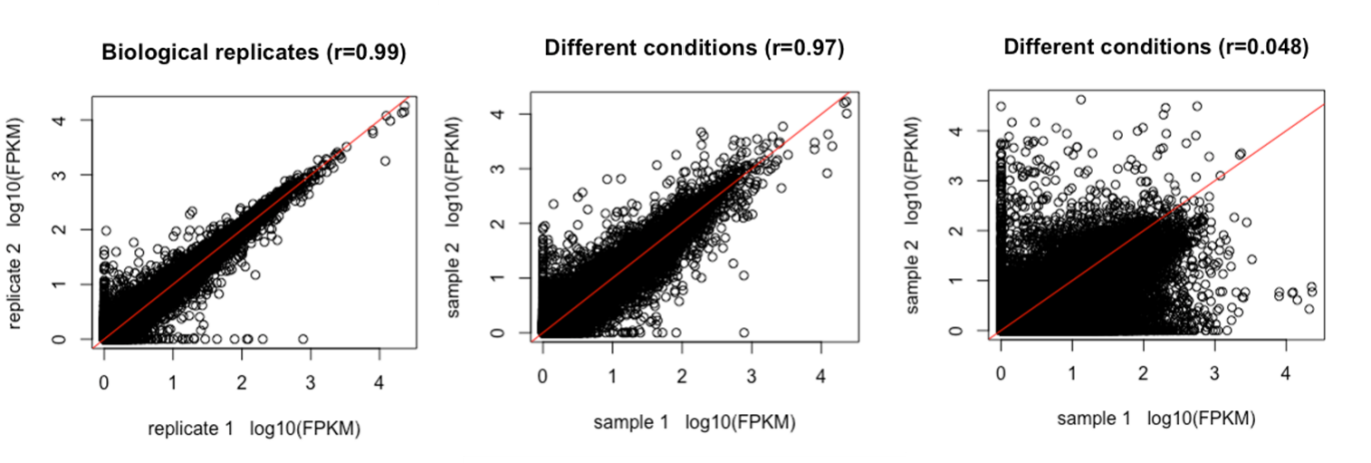
图:2 个重复样品(左)和两个条件(中和右)之间 FPKM 值的比较。重复之间的相关性应该非常高,两个条件之间的差异可以小(中)或大(右)。图片来源:CC BY-NC 4.0 [@own_5_2024]
可视化¶
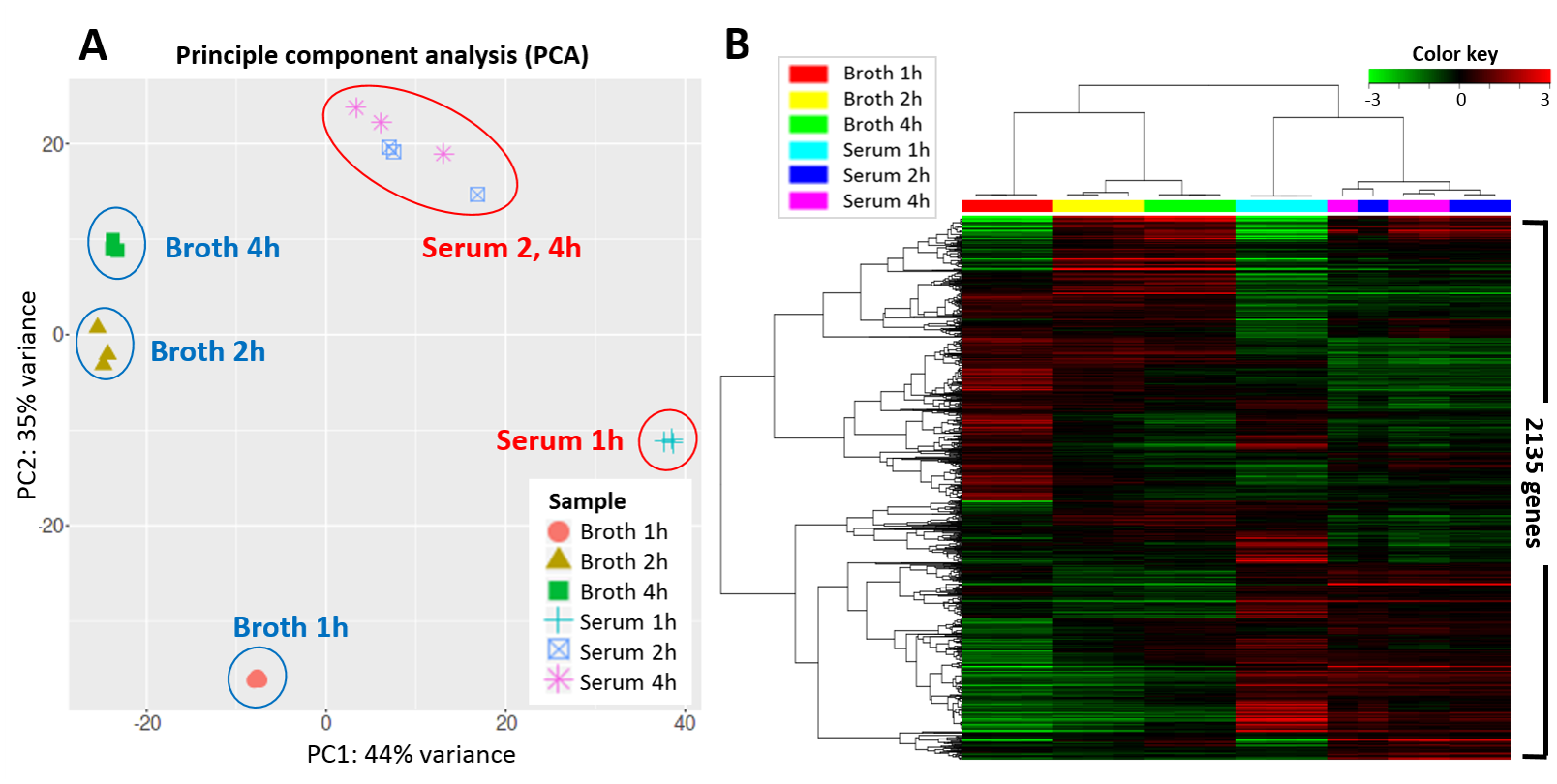
图:副乳房链球菌 (Streptococcus parauberis) 在两种不同培养基(鱼血清和肉汤)中生长 1、2 和 4 小时后 2,135 个基因的表达可视化。每个条件有 3 个重复测量。左图:主成分分析(PCA)显示两种培养基之间存在主要分离(44% 的方差),且有明显的随时间的进展。注意在血清上生长 2 小时和 4 小时后的表达差异不大。右图:热图可视化整个数据集,颜色表示 z-score 标准化的表达值:绿色为低表达,黑色为中等表达,红色为高表达。行为基因,列为生长条件,两者都进行了聚类。图片来源:CC BY 4.0 [@streptococcus_pca_heatmap_2021]
虽然组学数据可以在例如 Microsoft Excel 中查看,但很难从具有数万个基因和数十到数百个样品的数据矩阵中获得有意义的信息。因此,首先使用可视化或汇总数据的方法来查看是否可以检测到主要模式或异常值是明智的。一种广泛使用的可视化是所谓的热图——矩阵(基因 x 样品)的图像,每个测量值用颜色表示。如果数据沿基因和样品都进行了聚类,有趣的模式可能很容易被发现。初始数据探索中经常使用的第二种方法是主成分分析(PCA),它沿数据的主要变异轴绘制样品(或基因)。如果添加颜色或标记,PCA 图非常有助于检测分组和异常值。两种可视化方法都在上图中展示。
差异丰度分析¶
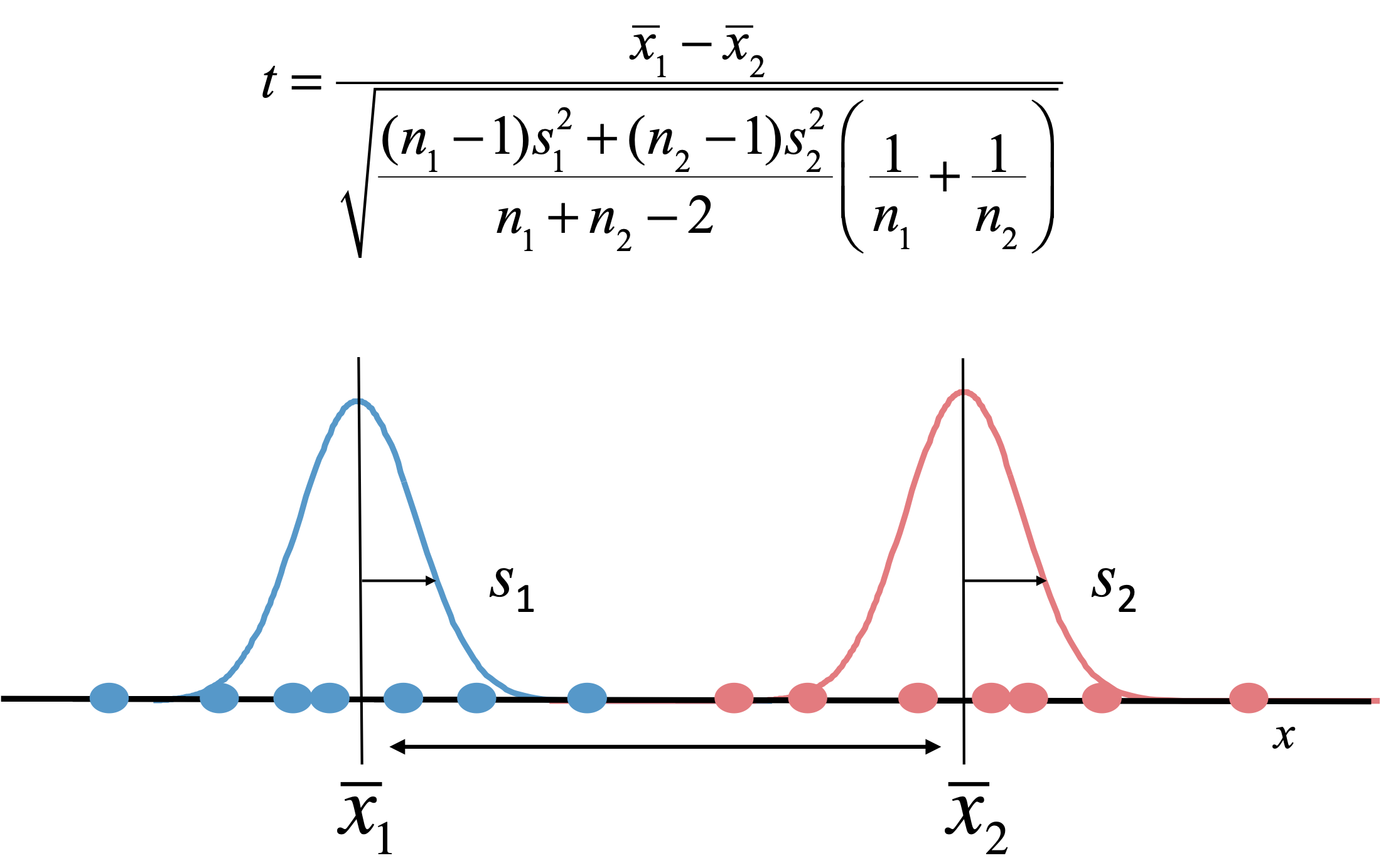
图:两个条件间基因差异丰度的最简单检验是 t 检验。t 统计量是两个分布均值 x 之间差异的度量,用标准差 s 表示的不确定性进行校正。p 值——我们偶然发现等于或大于当前 t 统计量的概率——可以使用 t 分布计算。图片来源:CC BY-NC 4.0 [@own_5_2024]
差异丰度分析是组学数据中最广泛使用的分析方法。目标是比较两类、条件、菌株、细胞类型等之间的丰度水平——例如健康与疾病组织、有无某种药物、不同生长条件等。最简单的方法是在两种条件下各收集若干重复测量,然后对每个基因进行简单的统计检验如 t 检验(见上图)。每个检验给出一个 p 值,p 值低于某个阈值(如 5%)的基因可以被称为显著差异表达。
有两个需要注意的问题:
-
如果你对数千个基因中的每一个进行单独检验,在 5% 的阈值下,你仍然会错误地将数百到数千个基因称为差异表达。为了解决这个问题,p 值通常经过多重检验校正,即被调大。
-
如果变异(上图中标准差 s)足够低,一个小的差异可以变得显著,即使实际丰度差异很小。因此,在许多实验中,选择基因的附加要求是倍数变化足够大。通常以 log2(倍数变化)表示,其中 +1 表示基因表达高 2 倍,+2 表示高 4 倍,-1 表示低 2 倍,以此类推。
有若干类似但更复杂的方法能更好地匹配实验后续分析,但这些超出了本书的范围。
时间序列分析¶
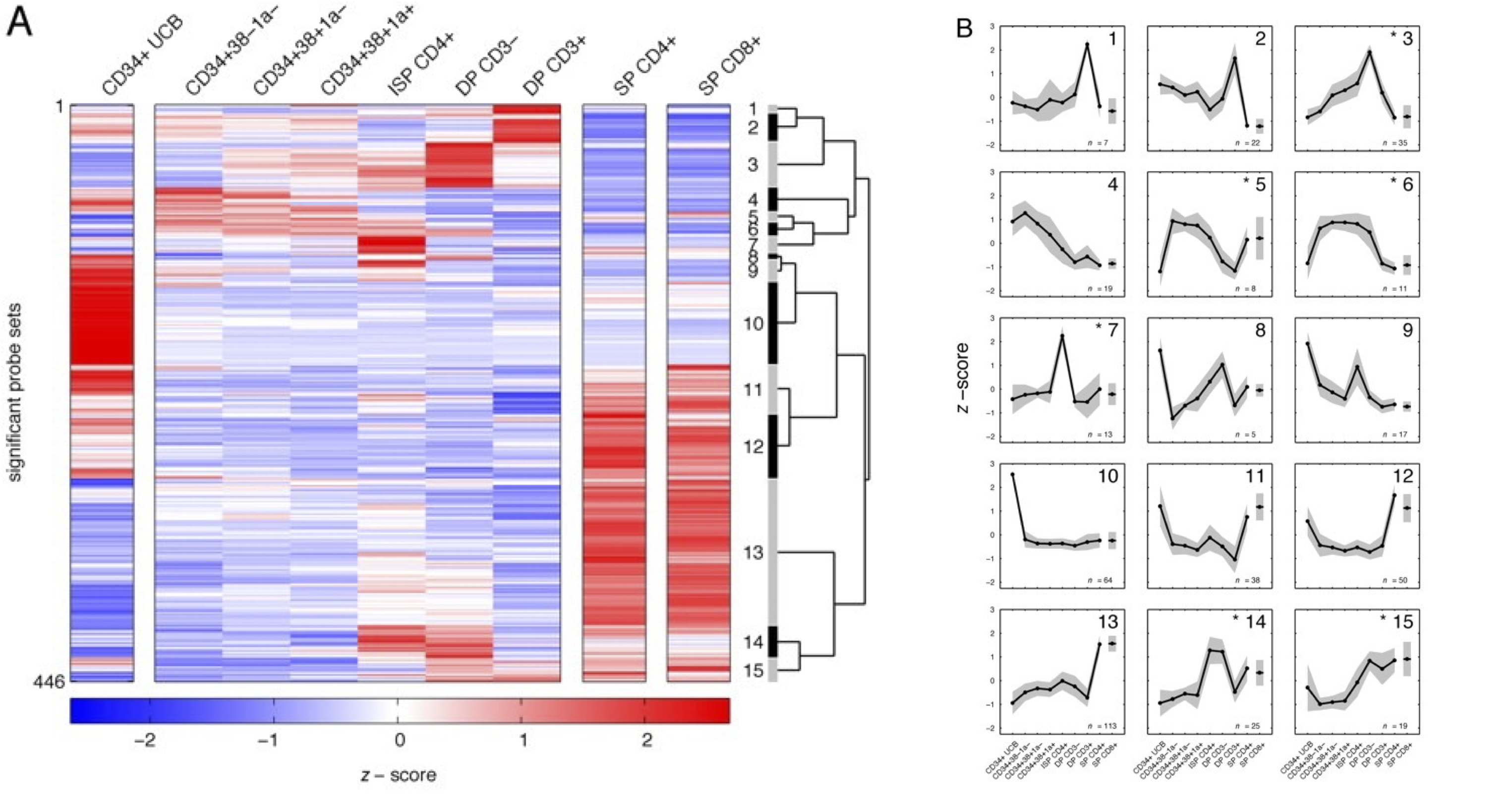
图:T 细胞发育各阶段的转录组学,即时间序列分析。左图:表达水平变化最大的 446 个基因的热图,聚类为 15 个簇。右图:每个簇的平均表达谱显示不同基因群在不同发育阶段达到表达峰值。这些基因可能以相同方式被调控,并参与相似的生物学过程。图片来源:CC BY-NC-SA 4.0 [@time_series_2005]
通常,跟踪丰度随时间的变化比在特定时间点比较更有意义,例如跟踪药物反应、生长条件变化、器官发育调控等。鉴于组学测量的成本,一个主要挑战是选择最佳采样时间点,平衡获得的信息与投入。后续分析包括聚类以找到相似调控的基因(见下文)以及更高级的方法——通过查看哪个基因的增加/减少先于另一个(组)基因来识别调控相互作用。上图提供了一个示例。
分类¶
分类与差异丰度分析相关,因为它试图找到最好地解释条件之间差异的基因。然而,这里的目标实际上是基于有限的一组基因表达水平尽可能准确地预测新(额外)样品的条件。应用主要在医学领域,如诊断和预后,但也用于区分不同细胞类型、发育阶段等。
聚类¶
聚类方法试图找到在所有样品上具有相似丰度谱的基因组,或者(反过来)在所有基因上具有相似丰度谱的样品组。我们称这些基因或样品为共表达的。基于"关联内疚"原则,相关性可以用于了解基因的功能——"如果基因 A 的表达与具有功能 F 的基因 B 相似,那么基因 A 可能也具有功能 F"。这有助于识别参与相似过程或通路的基因:共表达基因可能被共调控、编码相互作用蛋白等。对于样品,它可以帮助识别疾病亚型、不同基因型等,有助于了解不同的结果。聚类常用于排列热图的行和列(如上图和上上图),之后明显的聚类应作为大色块可见。
富集分析¶
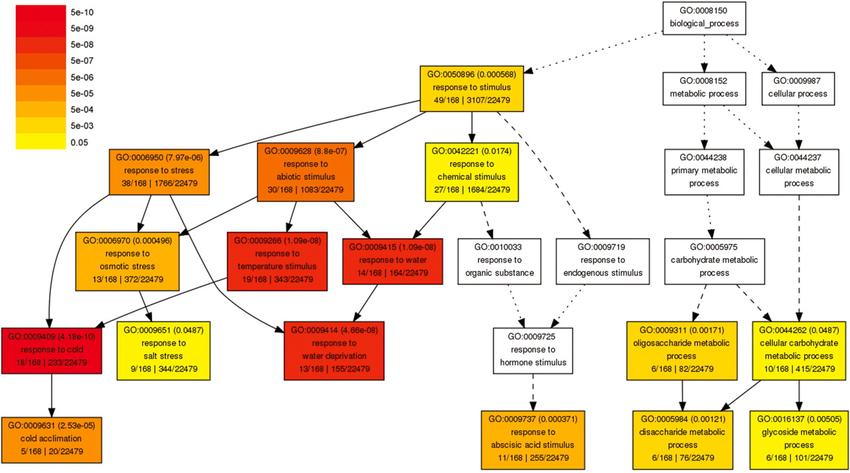
图:基因本体论(GO)的示例部分,在生物学过程类别中。低层级术语是高层级术语的特定实例。在此图中,GO 术语根据富集检验中的 p 值着色。图片来源:CC BY-NC 4.0 [@own_5_2024]
最后一种常用分析是富集分析,我们利用基因功能注释来了解一组基因,如差异表达基因列表或聚类。最广泛使用的基因注释是基因本体论 (Gene Ontology),一个结构化的术语字典,描述基因的分子功能及其在不同细节层次上参与生物学过程或细胞成分的情况(见上图)。然后统计检验评估我们在基因列表中发现特定注释的频率是否显著高于偶然预期。与差异丰度分析一样,产生的 p 值应进行多重检验校正。最终显著注释可以帮助在高于单个基因的层面解释实验结果。
展望¶
关于组学数据分析的这一节可能是最容易被淘汰的。我们仅触及或甚至省略了 DNA、RNA 和蛋白质单分子测量的最新发展、单细胞和空间组学分析(在单个细胞或组织网格点测量分子)以及深度学习的相关发展——这些深度学习方法承诺提供基础模型,利用大量的组学数据学习 DNA 和蛋白质的"语言"并解决特定任务。最终目标——活细胞的系统生物学模拟——仍远未实现,但可能比我们现在认为可能的更早实现。
实践作业¶
本实践包含问题和练习,帮助你处理第 5 章的学习材料。你有两个上午的时间来完成这些练习。在单个会话中,你应该目标完成本指南的大约一半(即第 1 天:作业 1-3,第 2 天:作业 4、5 和项目准备练习)。利用时间指示确保你不会卡在一个作业中。这些实践练习为你提供了最好的项目准备。特别是最后的**项目准备练习**很好地反映了撰写优秀项目报告所需的水平。确保你现在就发展实践技能,以便在项目中应用它们。
注意,答案将在实践结束后发布!
基因组学,20 分钟
使用下一代测序读段进行基因组组装由于读段短、测序过程中出现的错误以及基因组中重复序列的存在而具有挑战性。完成本练习后,你应该能够解释基因组组装的工作原理以及可能遇到的问题。
- 给定下面的 6 条短读段,尝试通过寻找读段之间的重叠来重建原始 DNA 序列。注意可能存在一些测序错误。该 DNA 序列编码的氨基酸序列是什么?提示:将序列复制到 Word 中使用等宽字体(如 Courier),并添加空格对齐序列;对于翻译,可以在 Google 上搜索在线工具。
>read_1
aagcagcgcgggcgaataataataa
>read_2
acccatattagcattagcacccttg
>read_3
cccatgaacatattgatgatgaaaa
>read_4
aaaacatggaaagcagcgcgggcga
>read_5
ttagcattagcacccatgaacatat
>read_6
gatgatgcaaacatggaaagcagcg
- 如果没有 read_6,你能明确重建整体序列吗?
基因组学与 NGS,60 分钟
在这个简短的实践教程中,你将使用基因组浏览器来了解特定基因及其序列变异;然后你将使用 IGV(Integrated Genome Viewer)来检查多个不同的 NGS 数据集。请注意,在下面讨论的大多数环境中,你可以通过将鼠标悬停在图表元素上(箱线图、点等)获取更多信息。请使用 Google Chrome 或 Firefox,因为其他浏览器可能无法很好地显示所有网站。
完成本练习后,你应该能够解释下一代测序数据如何用于研究基因组保守性、遗传变异和基因组功能。
-
本基因组浏览器介绍将基于一个叫做 BCL11B 的基因。首先在 GeneCards 数据库 中查找有关该基因的信息。它是如何得名的?
-
访问 UCSC Genome Browser,进入 Genome Browser("Tools" 下方)。在 "Human Assembly" 下选择 "hg19" 基因组组装并搜索 BCL11B。如果你得到可能位置的列表,请选择对应 BCL11B 基因的正确链接。该基因在哪条染色体上,长度是多少(以 bp 计),有多少个外显子?该基因从什么位置开始(提示:检查基因模型中的箭头)?
-
点击图面板顶部的基因变异,访问包含基因模型信息的页面。编码区域有多长?ORF 有多长(以 bp 和氨基酸计)?
-
返回基因组视图。基因组浏览器使用 "tracks",即与基因组对齐的各种信息来源。"100 Vert. cons" 轨道包含什么?你是否看到该轨道与另一个轨道之间的相关性?
-
你也可以自己向视图中添加轨道。在主基因组窗口下方,查找(但暂时不要点击)"Phenotype and Literature" 下的 "ClinVar Variants",使用下面的下拉菜单选择 "pack",然后按右侧的 "refresh" 按钮。ClinVar 是一个包含已证明临床效应(即导致疾病)突变的数据库。找到第四个外显子中的红色 T>G SNP——在 "ClinVar Short Variants" 下——并放大到核苷酸水平('base'),检查它是否为同义突变(提示:使用顶部的 "--->" 选择正确的链)。这是否符合你的预期?
-
该位置在其他基因组中是否保守?
你可以在 UCSC Genome Browser 中检查大量预计算的轨道,但很难可视化单个 NGS 样品。为此,我们转到 IGV——Integrated Genome Viewer。查看文档(顶部菜单中的 "Help")以熟悉用户界面。
熟悉 IGV 后,首先在 "Genomes" 菜单项下选择 hg19 人类基因组组装。然后使用搜索框查找 BCL11B 并加载一些额外的轨道。首先,选择 "Tracks -> Variants",然后从列表中选择 "1KG Phase 3 SNPs";这些是在 1000 人类基因组项目中发现的基因型(该项目最终测序了约 2500 个基因组)。另外选择 "Tracks -> Platinum Genomes" 并选择 "NA12878";这是使用不同技术深度测序的六个基因组之一的短读段数据,具体来说是一个淋巴母细胞系。
-
放大到(左侧的)最后一个外显子。你在 NA12878 读段数据和该轨道顶部的覆盖度图中注意到了什么?
-
放大到位置 99,639,270。1KG 基因型轨道告诉你该位置的什么信息?点击轨道元素获取更多信息。
-
NA12878 在该位置有什么基因型?有多少条读段支持该基因型?
-
接下来,从 ENCODE 项目加载调控转录数据。ENCODE 项目是一个大型功能基因组学项目,提供了关于人类基因组如何使用的各种数据集:转录活性、组蛋白修饰、DNA 可及性等。下载 NA12878 的 RNAseq 数据文件 并使用 "Tracks -> Local File..." 在 IGV Web 应用中作为轨道加载。如果你仔细观察 BCL11B 全长的这个轨道,它是否与基因模型一致?什么可能导致不一致?
结构变异是基因组中可以检查的另一类变异。
-
转到 chr1:85,974,000-85,993,000(将坐标复制粘贴到搜索框中)。仔细查看 NA12878 的读段数据轨道。读段覆盖度暗示这里有什么类型的结构变异?查看该论文中的 Figure 3A。这是否与你的观察相符?从 "Tracks -> Variants..." 菜单加载 "1KG Phase 3 SVs" 来验证这一点。
-
定位结构变异的另一端。它的大小是多少?
-
在 IGV 中,一个基因与结构变异重叠。该基因的名称是什么?你认为结构变异是否影响该基因的蛋白质序列?
- 你可以使用 "Tracks -> ENCODE Signals..." 可视化 ENCODE 项目中生成的各个数据集。在右上角框中搜索 "12878" 并选择几个轨道来显示。使用 Google 了解这些轨道中显示的是什么实验。
这就结束了基因组浏览器的试用,展示了你可以如何相对容易地检查基因组数据中的保守性、变异、表达等。虽然这里我们聚焦于人类基因组(因为数据的广泛可用性),IGV 可以处理任何基因组数据——只要它是标准格式——且类似于人类 UCSC 的基因组浏览器在 UCSC、EMBL-EBI 和 Ensembl Genomes 以及模式生物社区站点上可用于许多不同生物体。
组学数据分析,45 分钟
分析组学数据的工具有多种多样;本书无法介绍所有这些工具。为获得一些经验,我们将探索一个预处理的人类癌细胞系转录组学测量在线合集——癌症细胞系百科全书(CCLE)。数据托管在 Broad Institute(哈佛和 MIT 之间的合作)的 Cancer Dependency Portal (DepMap) 上,作为 Broad Cancer Dependency Map 的一部分。
CCLE 项目存储了使用 RNAseq 测量的所有人类基因的表达。RNAseq 测量值以 log2(TPM+1) 表示,其中 TPM 代表每百万转录本数(每个基因的标准化读段计数)。该项目还包括拷贝数(CN)变异、DNA 甲基化等测量。
再次提醒,请使用 Google Chrome 或 Firefox,不要忘记在大多数工具中你可以通过将鼠标悬停在图表元素上来获取更多信息。完成本练习后,你应该能够解释可以对定量数据执行的主要分析类型(差异表达、聚类、富集分析)。
-
访问 CCLE 网站并搜索我们之前使用的基因 BCL11B。你的初始视图包含大量关于该基因在不同(患病)组织中表达和突变的信息。选择顶部的 "Characterization" 标签。然后你会看到 BCL11B 基因的表达,使用所谓的箱线图进行可视化。使用 Google 了解如何解释这些图——箱体和线代表什么?
-
在左侧菜单中选择 "Show lineage subtypes" 框。该基因在哪些组织/肿瘤中高表达?在这些组织中,该基因在哪些疾病中低表达(即在哪些疾病之间 BCL11B 差异表达)?这与你在 GeneCards 中了解到的信息一致吗?
-
通过点击 "Explore relationships with other data"(右上角),你可以更深入地探索数据。在初始视图中,y 轴仅用于分隔数据点,它实际上不显示测量值。将鼠标悬停在点上以获取关于测量 BCL11B 的样品的更多信息。通过在 "View Options" 下为 "Color by" 选项选择 "Model Property",然后选择 "Primary disease" 作为属性,可以获得更具信息量的图。在哪些疾病和样品中你发现最高表达?
-
现在将 "Plot type" 设为 "Scatter plot"(左上角)。然后对于 Y 轴,在 "Data type" 下选择 "Expression",在 "Feature" 下选择 "Gene" 和 "BCL11A"。如果需要,在 "View options" 下选择按 "Primary disease" 着色。现在每个点代表一个样品中 BCL11A 和 BCL11B 的表达。在哪些疾病类型中两个基因都高表达?
-
现在以相同方式比较 BCL11B 与基因 RHOH 的表达。你看到了什么?你可以再次将鼠标悬停在点上以获取更多信息;在右侧的图例中,你也可以双击特定疾病以隔离单个疾病并获得更清晰的图。
-
关于 RHOH 本身的**差异表达**你能说些什么?如有需要,请尝试 "Group by" 和 "Filter by" 视图选项。差异表达模式合理吗?你可以通过 Google 搜索这些疾病来了解更多关于它们发生的组织。
-
在 GTEx 中查找 BCL11B——基因-组织表达合集。GTEx 类似于 CCLE,但在健康组织中分析表达,从捐赠者(在去世后直接)收集。在 "Expression" 菜单项下搜索 BCL11B 基因。在生成的图中,通过交互操作找出 BCL11B 在哪个组织中通常表达最高。
-
在 GTEx 中,你可以检查样品的一些 PCA 图。PCA(即**主成分分析**)常用于可视化高维数据。它找到样品在数据中差异最大的方向:PC1 是变异最大的方向,PC2 是垂直于 PC1 的下一个方向,以此类推。使用顶部菜单,进入 "Expression" -> "Expression PCA"。选择 "Hardy Scale" 为样品着色。你认为这个量表衡量什么?提示:查看图下方的图例,颜色对应 Hardy Scale。
-
你期望在哪个组织中看到 "呼吸机病例" 捐赠者的表达差异?从 "Select a tissue" 下拉框中选择该组织以创建组织特异性的 PCA 图来验证这一点。
-
返回 CCLE 并绘制 BCL11B 表达与 BCL11B 蛋白质组学的对比图。你预期存在相关性吗,你是否看到了?
-
绘制 BCL11B 表达与其甲基化的对比图,并检查 DNA 甲基化的作用 (https://en.wikipedia.org/wiki/DNA_methylation)。结果是预期的那样吗?
聚类,30 分钟
如果一切顺利,你应该在某些组织中发现了 BCL11B 和 RHOH 表达之间的相关性。通过执行聚类——即找到具有相似表达的基因组和/或样品组,可以更容易地发现此类关系。此类聚类可以帮助你了解聚集在一起的基因之间的功能关系,或聚集在一起的样品之间的表型关系。对于 CCLE,有一个很好的交互式查看器可在这里使用。打开页面并选择 "Bone" 子集。
-
在主图中,所谓的**热图**,蓝色和红色像素对应什么?注意你可以使用鼠标放大和滚动;并阅读图下方的文字。
-
BCL11B 在样品 RDES 中的表达水平是多少(如需要可使用搜索框)?
-
在右侧和底部,聚类由灰色条指示。你最初看到多少个基因组?多少个样品组?考虑到基因表达值,你认为这合理吗?
-
你可以使用聚类旁边的 "volume slider buttons" 创建更少或更多的聚类。试试这些。将基因分成更多聚类看起来合理吗?样品呢?
-
放大左侧的基因名称。BCL11B 确实与 RHOH 聚在一起了吗?
-
在顶部,显示了关于样品的额外信息(组织、组织学等)。其中一个样品簇是否对应某个注释?
-
左上角显示了若干基因本体论术语(回忆第 1 章),这些术语在骨肿瘤相关基因集合中出现的频率高于随机预期(即比从所有基因中随机抽取相似大小的基因集合时更频繁)。我们称之为 GO 富集分析。条的高度对应于该分析的 p 值;条越高,富集越显著。这些术语有意义吗?
差异基因表达,45 分钟
NCBI 托管着基因表达综合数据库 (Gene Expression Omnibus),包含基因表达实验——包括微阵列和 RNAseq。你可以以各种格式下载和组合这些数据,并在自己的计算机上进行分析,可能在不进行自己实验的情况下回答生物学问题。本课程时间太短,无法教授重新分析公开数据所需的所有技能,但我们可以使用一个名为 GEO2R 的在线工具在 Web 浏览器中进行一些简单分析。
完成本练习后,你应该能够解释定量数据差异表达分析的结果。
-
访问 Gene Expression Omnibus,搜索样本系列 GSE69485(微阵列数据)并阅读产生该系列的研究摘要和设计。在底部你可以找到样本列表。有多少个?
-
使用的是什么类型的微阵列?你可以在 GEO 中搜索 GPL90 获取更多信息。有多少个探针集(对应于数据表中的行数)?对应多少个基因?
你可以在 GEO2R 中对这些数据进行简单分析。在这个案例中,我们将尝试找到在需氧和厌氧条件下生长的酵母之间差异表达的基因,即有氧与无氧。要开始,返回样本系列 GSE69485 的页面,按页面底部的 "Analyze with GEO2R" 按钮。你将看到所有样本的列表,你现在可以将它们分配到不同组。首先点击 "Define groups"(在顶部)创建两个组,比如 "aerobic" 和 "anaerobic"。然后选择厌氧条件下指数生长期(包括晚期/末期)采集的四个样品并将它们分配到厌氧组,通过点击组列表中的 "anaerobic"。对四个有氧指数生长期样品做同样的操作。
- 向下滚动并按 "Analyze" 按钮。经过一些处理后,显示了若干图表,呈现表达水平、倍数变化、差异表达基因(蓝色/红色)等。"expression density" 图包含每个样品中所有基因表达值的总体分布;如果这些分布看起来非常不同(例如由于测量设置的差异),这可能暗示问题。这些阵列看起来可以比较吗?
在图表下方,你应该看到按(校正后的)p 值排序的 Affymetrix 探针集列表(在 "ID" 下)。logFC 列表示 log2 倍数变化:0 表示无变化,正值表示在厌氧条件下更高表达,负值表示在有氧条件下更高表达(反之亦然,取决于你先创建哪个组)。
-
这里最显著的差异表达基因是什么(p 值最低的)?
-
你可以通过点击表格中对应的行来查看底层表达值,这将显示每个样品中的表达图;"Sample values" 按钮将显示实际测量值。比较前三个基因的表达范围。你注意到了什么?
-
要了解更多关于差异表达基因的信息,你可以添加基因本体论(GO)信息。为此,点击表格上方的 "Select columns" 并选择 "GO: Function"、"GO: Process" 和 "GO: Component"。然后按 "Set"。你注意到前 20 个基因中有什么类型的功能?
理想情况下,获得的结果应通过额外的实验加以验证。在 GEO 中,另一项关于在酵母中工程化淀粉酶基因影响的研究可用,产生了系列 GSE38848。
- 在系列 GSE38848 上使用 GEO2R 找出仅在参考菌株(NC)中厌氧和有氧条件下差异表达的基因。你找到了相同的基因吗?相同的功能?
注意,这只是使用若干工具解读组学数据的快速导览,针对一些非常特定的数据集(人类癌细胞和酵母细胞中的转录组学)。然而,底层的分析——差异表达、聚类、主成分分析等——可以广泛应用于大多数定量组学数据集。如果你想深入研究,需要使用更高级的(统计)方法,这些在其他课程中教授。
项目准备练习
探索 ARF5 和 IAA5 的组织特异性基因表达。为此,你可以使用一些其他(植物特异的)资源,而不是上面使用的人类为中心的资源,例如 Expression Atlas(也可通过 UniProt 在 "Expression" 下访问)或 BAR——植物生物学的生物分析资源。
用几条要点分别描述以下项目。你可以包含最多两个图表或表格。
-
材料与方法 你做了什么?你使用了哪些数据、数据库和工具,为什么选择这些?你选择了哪些重要设置?
-
结果 你发现了什么,主要结果是什么?报告相关数据、数字、表格/图表,并清楚地描述你的观察结果。
-
讨论与结论 结果合理吗?是否符合你的预期,还是你看到了令人惊讶的东西?结果意味着什么,你如何解释它们?不同工具是否一致?你能得出什么结论?确保描述你的解释所基于的预期和假设。
附录:组学测量技术¶
我们现在对细胞分子生物学的了解大多基于广泛的测量,使用了不断改进的一系列设备。测量数据的数量及其覆盖度、可靠性、偏差等关键取决于这些设备背后的技术及其局限性。此外,(缺乏)技术影响我们目前还无法测量的内容,因此我们对这些方面相对"盲"。这是在分析所得数据时需要牢记的最重要的信息。出于这个原因,我们在正文中只列出了主要的数据特征。这里,我们为感兴趣的读者提供关于最重要(历史性)技术的更多背景。
基因组学¶
知识框 5.8:Sanger 测序
由 Fred Sanger 及同事于 1977 年开发,该方案最初主要是手动操作,直到 1985 年被 Applied Biosystems 自动化。Sanger 测序使用链终止法(见下图)。
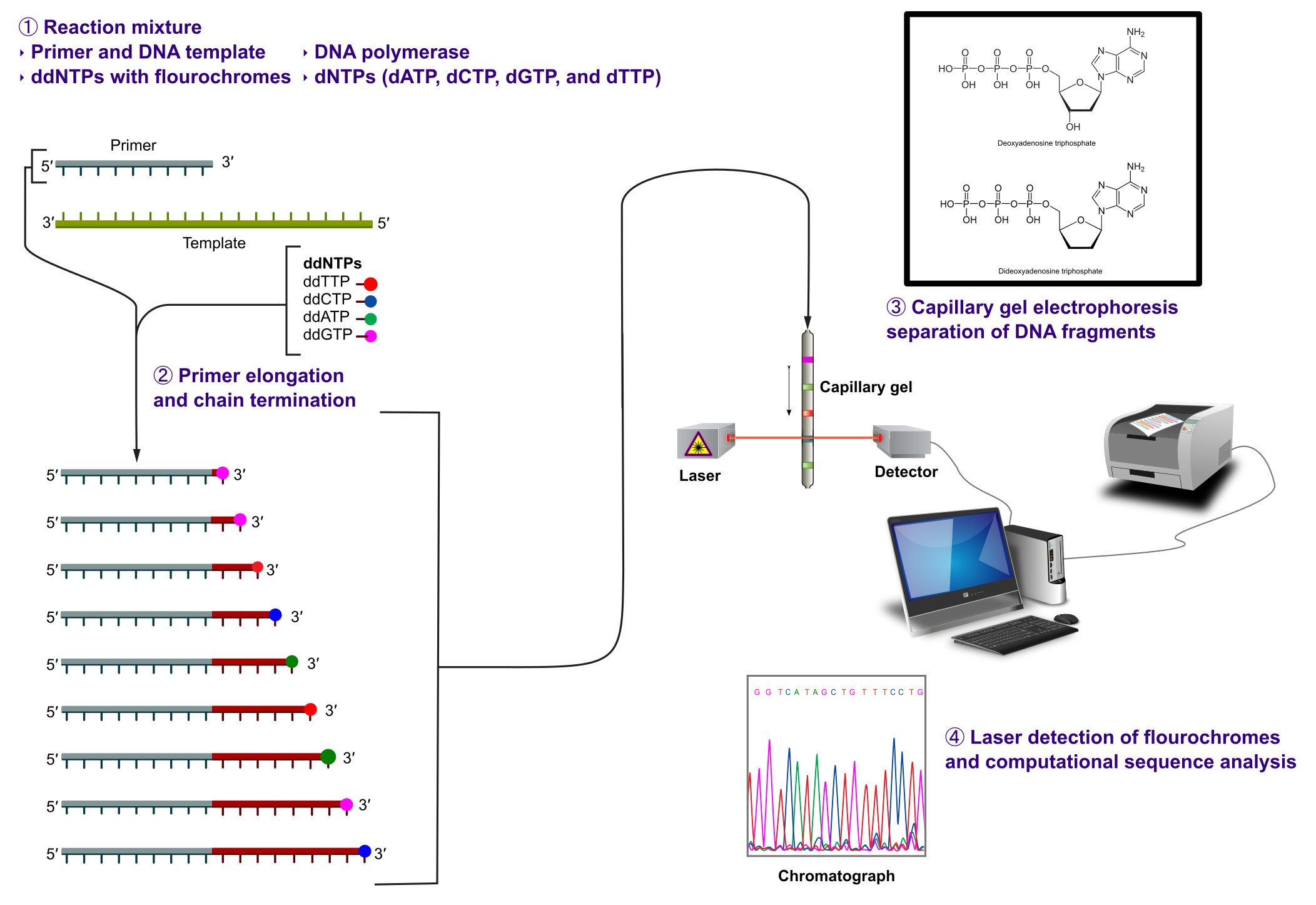
图:链终止(Sanger)测序。图片来源:CC BY-SA 3.0 [@sanger_2012]
在第一步中,基因组 DNA 被纯化并打碎为所需长度的片段。对于 Sanger 测序,片段长度约为 1,000 个核苷酸。打碎通过机械或化学方式进行,所得片段长度并非完全相等:它们围绕目标片段大小分布。
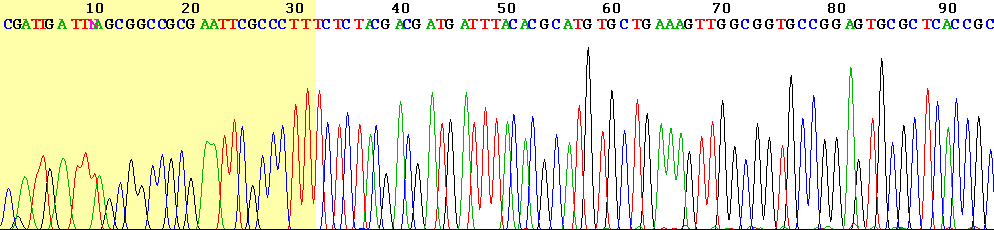
图:Sanger 测序信号,读段起始处有低质量碱基。图片来源:CC0 1.0 [@sanger_signal_2005]
模板 DNA 片段在 PCR 反应中使用引物和 DNA 聚合酶进行扩增。每个样品在 4 个独立反应中扩增,每个核苷酸(A、T、C、G)一个。在每个反应中,添加少量修饰核苷酸(ddNTP)到正常核苷酸(dNTP)中。这些修饰核苷酸设计用于终止链的延伸,并连接一个可以识别的标记。这产生了一部分扩增的模板 DNA 片段集合。每个片段的长度最初通过片段在凝胶上的迁移距离来测量,在后来的设置中通过通过毛细管的时间来测量。最后一个核苷酸上的标记然后识别给定位置的碱基,并生成峰值模式。从峰值模式的信号(见上图),序列可以自动读取。Sanger 测序产生 700-1,000 个核苷酸的读段长度;在此之后,碱基识别质量下降太多而无法使用。读段起始处的质量通常太低而无法使用(见上图)。Sanger 测序的另一个问题是同聚物(同一核苷酸连续出现多次)的检测,因为信号的峰值高度随延伸长度而降低。这使得难以区分 3、4 和 5 个相同碱基。Sanger 测序机可以并行测序 96 个片段,使其通量相对较低。
知识框 5.9:Illumina 测序
Illumina 测序使用桥式扩增,其中 PCR 反应直接在流动池表面进行。在文库制备步骤中,正向和反向接头被连接到单链模板片段的两端。接头的互补序列作为 PCR 引物连接到流动池上。初始模板序列(含接头)将与流动池表面的一个引物形成双链键。然后片段在标准 PCR 反应中被复制,但末端牢固地附着在表面上。DNA 被变性从双链回到单链,原始模板被洗掉,留下一个固定的单拷贝。该链的末端然后可以与另一个末端的邻近空引物形成桥,反应重复进行,最终产生两个固定片段,随后在该片段附近产生数千个相同的片段。在测序步骤中,最后的 PCR 使用荧光染料终止的 NTP,在每个循环中被冲洗过表面。相机检测颜色,染料被切除,步骤重复进行测序反应的长度:
当前型号使用所谓的图形化流动池,模板片段落入微小孔中,扩增步骤在孔中进行:
知识框 5.10:PacBio 测序
PacBio 测序不在测序前扩增模板片段,而是巧妙利用 SMRT-cell 的结构,通过激光放大碱基掺入时的光信号。环化的双链模板被加载到 SMRT-cell 上的微小孔中,目标是在每个孔中只有一个分子。核苷酸的掺入通过从核苷酸上切下一个磷光分子来发出信号,并用相机记录。
与 NGS 的一个主要区别是模板是环化的而非线性的,因此单个模板可以被连续测序多次,这被称为环形共识测序(CCS)。一般来说,第三代测序技术具有较高的错误率,大多数错误为 indel(短插入或缺失)。这对映射和组装有影响。利用 CCS 允许校读和更高准确率,最新的 PacBio HiFi 读段达到 99.99% 的读段准确率(Q40)。
该技术的读段长度由输入片段大小和聚合酶功能持续时间决定(高能激光光随时间缓慢降解酶)。CCS 读段的中位读段长度在 15,000 到 20,000 个核苷酸之间,连续长读段测序最长可达 175,000 个核苷酸。PacBio 测序没有其他技术那样的扩增偏差(没有 PCR 步骤),受 GC 含量的影响最小。总体而言,它在基因组序列上给出最均匀的覆盖度。遗憾的是,它的通量远低于 Illumina 测序,每碱基成本仍然显著更高。
知识框 5.11:Nanopore 测序
纳米孔测序的流动池有若干孔。每个孔底部有一个检测电流的传感器。孔本身由膜覆盖,类似于细胞膜,尽管在这种情况下它不是脂质双层而是更稳定的聚合物。膜中嵌有跨膜蛋白孔(经过基因工程改造以优化功能),DNA 分子可以穿过。在膜的顶部和底部施加电流势,由于 DNA 带负电,它想要穿过孔。这改变了电阻,被传感器检测到。一个问题是 DNA 穿过速度太快,传感器无法检测核苷酸;解决方案是添加 DNA 聚合酶作为"刹车"(它也被称为马达,因为它主动解旋 DNA)。聚合酶本身不能穿过孔,而是坐在孔的顶部。
与 PacBio 一样,读段长度由输入 DNA 片段大小决定,没有理论限制:当前的读段长度记录为 4.2 Mb,足以在单次读段中测序一个细菌基因组。纳米孔测序具有与 PacBio 类似的错误模型,插入和缺失最为常见。除此之外,长同聚物延伸仍然具有挑战性。准确率限制在 96.8-99.7%(Q15-Q25)之间。相对于所有其他技术的主要优势是直接测量模板 DNA 本身,因此也可以检测碱基修饰如 DNA 甲基化。测序仪也足够小可以放在手掌中,人们已在各种非传统条件下使用它,如在斯瓦尔巴群岛的北极探险和国际空间站中。
转录组学¶
知识框 5.12:凝胶与 qPCR
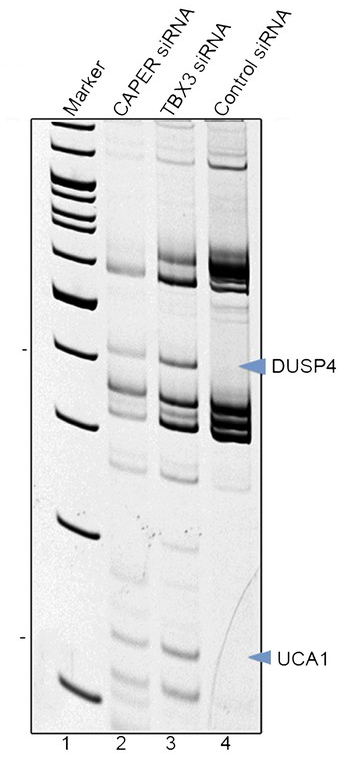
图:差异显示凝胶的示例。图片来源:CC BY 3.0 修改自 [@differential_gel_alt_2014]
早期检测转录本和表达水平的方法是 Northern 印迹和差异显示(见上图)。两者都是基于凝胶的方法,通量低且不太准确。Northern 印迹和差异显示被 qPCR(定量 PCR)和微阵列所取代(见知识框 5.13)。qPCR 是一种 PCR 形式,通过添加荧光报告子(结合 DNA 的染料或荧光探针)来测量每个 DNA 分子的丰度。荧光水平随扩增片段数量的增加而增加,从而被检测到。当反应在给定循环中超过阈值时,循环数用于推断反应中原始模板片段的数量(见下图)。qPCR 常用于验证其他定量方法获得的结果。
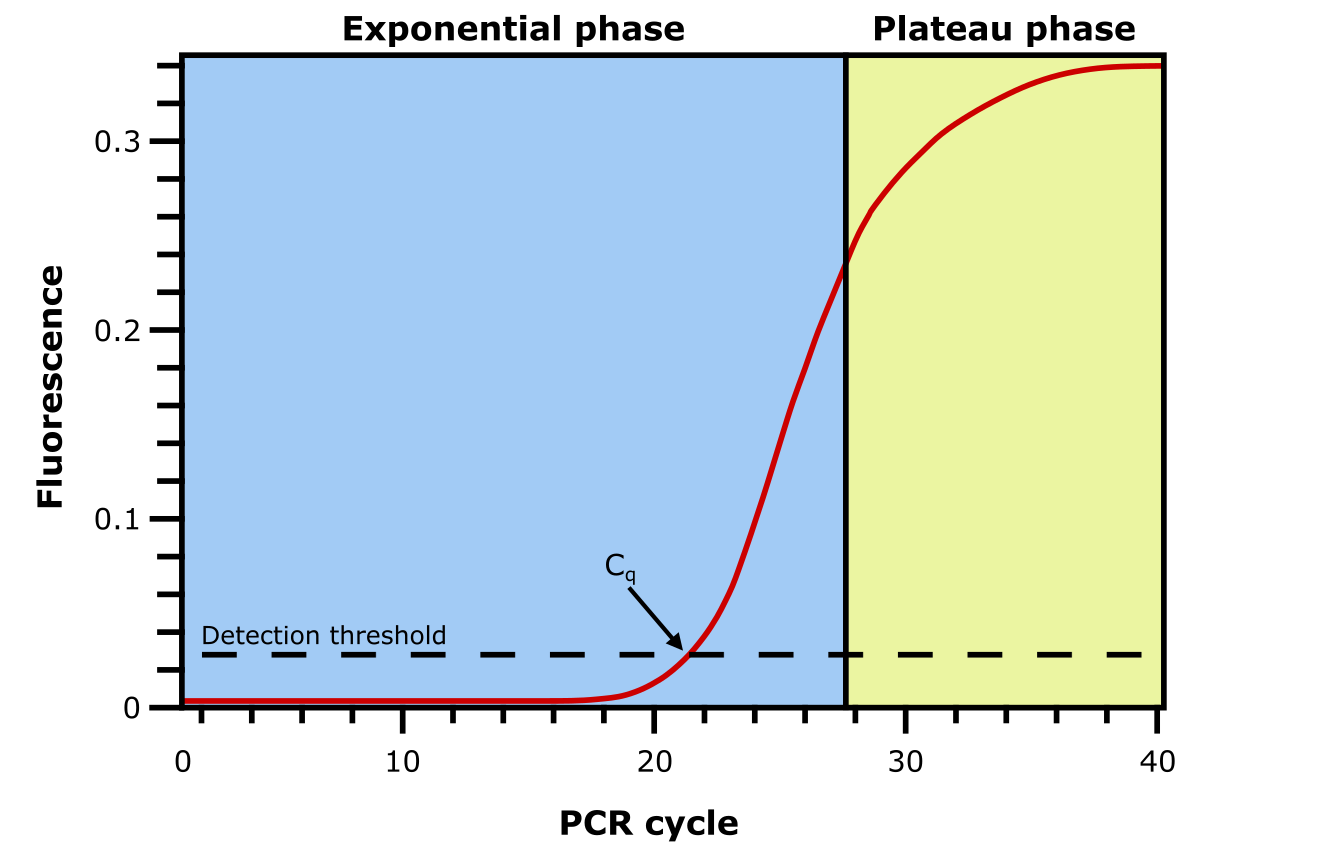
图:qPCR 反应中 DNA 片段的扩增图。Cq 对应荧光通过检测阈值的循环。图片来源:CC BY-NC 4.0 [@own_5_2024]
知识框 5.13:微阵列
第一种广泛使用的测量基因表达的高通量方法是微阵列。DNA 微阵列基于互补 DNA 链相互结合的原理。微阵列通常是平坦表面(玻璃片或其他材料),上面有已知位置的微小单链 DNA 序列——所谓的探针——从几千到数百万个不等。每个 DNA 序列被选择(尽可能地)代表特定基因,即一个独特子序列。这意味着微阵列只能设计来检测已知基因,且是物种特异的,基因变异(SNP、剪接变异体)很难检测。
在微阵列实验中,首先通过寻找 poly-A 尾来选择 mRNA 分子,转换为互补 DNA(cDNA),用荧光染料标记,然后冲洗过表面。互补序列会结合,一段时间后未结合的材料被洗掉,使用显微镜测量荧光。表面上特定位置的光强度水平是结合序列数量的间接读出,因此反映相应基因的相对表达。
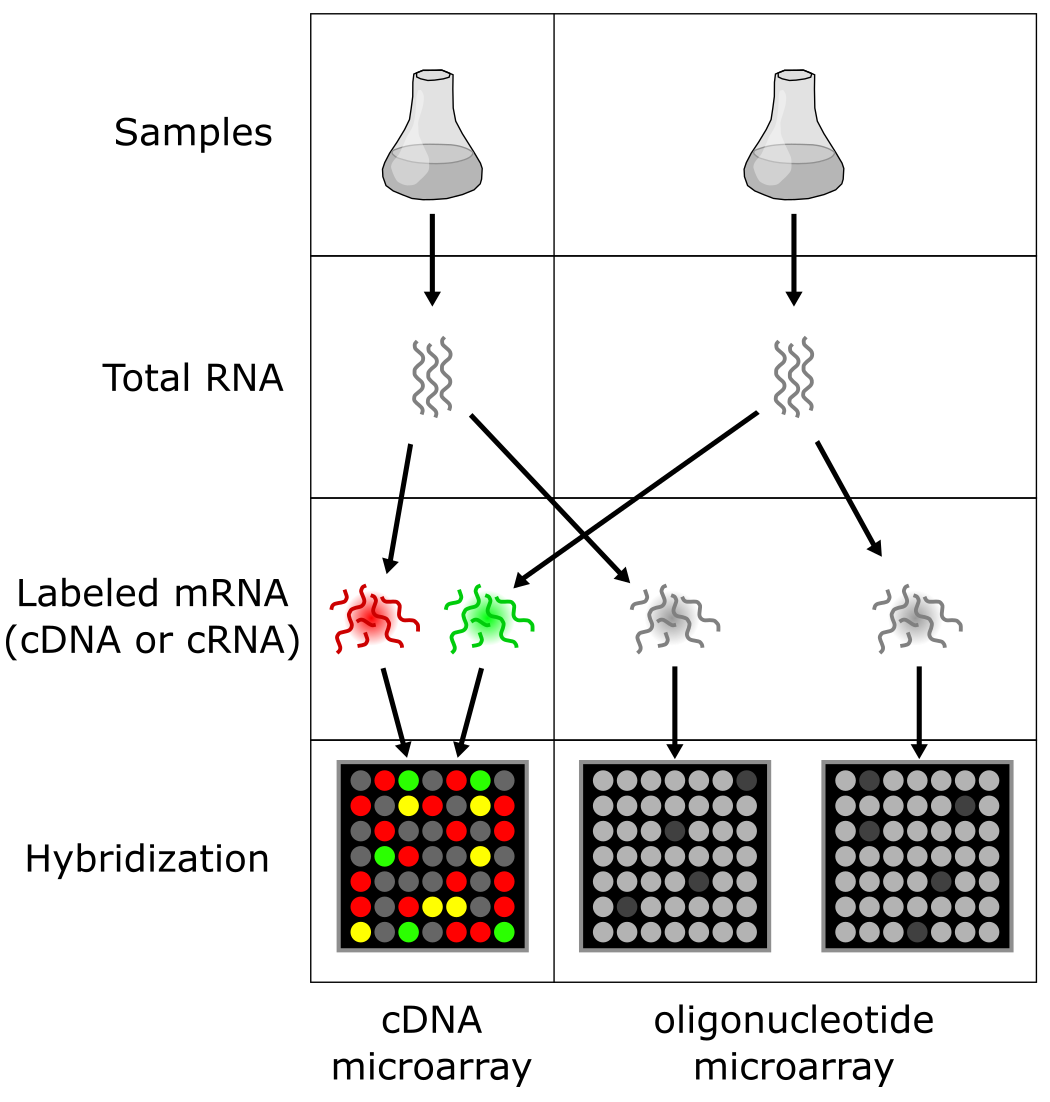
图:cDNA(双色)与寡核苷酸(单色)微阵列分析的区别。图片来源:CC BY-NC 4.0 [@own_5_2024]
有两种主要的竞争性微阵列类型:cDNA 和寡核苷酸阵列。虽然原理相同,但它们在生产和使用上有所不同(如下图所示):
-
**cDNA 微阵列**含有长探针,从几百个核苷酸到 1000nt。它们可以在实验室中通过点样机器人生产,因此可以轻松为特定实验进行调整。这带来了微阵列之间的更大变异,使得比较不同测量更加困难。cDNA 微阵列因此主要用于两个样品之间的直接比较,其中两个样品(例如健康和疾病组织)用不同的荧光素标记——通常是 Cy3(绿色)和 Cy5(红色)。结合的 DNA 序列的相对数量反映 mRNA 分子在样品 1 和样品 2 中的相对浓度,因此反映颜色。绿色斑点意味着只有样品 1 包含相应的 mRNA 分子,红色斑点意味着只在样品 2 中找到,黄色斑点意味着在两个样品中都找到了。强度反映总体表达水平:两个样品中都低表达为黑色/暗色,高表达为亮色。
-
**寡核苷酸微阵列**含有短探针(25nt),使用类似于微芯片生产的技术生产。这意味着质量高且恒定,不同阵列可以轻松比较。寡核苷酸阵列因此通常只使用一种颜色测量单个样品。然而,由于短探针不太可能对基因唯一,转录本通常通过组合多个探针到所谓的探针集中来测量。
总体而言,微阵列测量往往有噪声,无法区分非常低的表达水平,因为它们不提供足够的荧光信号。因此数据标准化也是一个重要步骤,以去除不同微阵列测量之间的非相关变异。
蛋白质组学与代谢组学¶
知识框 5.14:质谱扩展技术
质谱扩展技术¶
质谱仪可以提供相当精确的 m/z 测量,但仍然有许多分子具有(几乎)相同的质量,因此无法使用单个 MS 设备区分。因此广泛使用若干扩展方法来基于其他标准分离分子:
-
凝胶电泳:与 DNA 和 RNA 一样,凝胶可用于基于大小和电荷分离其他分子。之后,可以切出特定条带,用 MS 设备测量分子含量。凝胶位置和质谱的结合可以帮助缩小实际存在的确切分子范围。最广泛使用的凝胶类型是 2D-PAGE(聚丙烯酰胺凝胶电泳),按等电点和分子量分离分子。
-
色谱法:在进入 MS 设备之前,通过分离柱移动样品——填充惰性气体(气相色谱法,GC-MS)或液体(液相色谱法,LC-MS)。不同分子通过这些柱的时间不同,到达 MS 设备的时间因此提供额外信息。概念类似于使用凝胶,但柱可以直接连接到 MS 设备以实现连续通量。
-
串联质谱或 MS/MS:分子被测量两次——先完整测量(在第一个 MS 设备中),然后在选择和碎裂后再次测量(在第二个 MS 设备中)。这依赖于碎裂的可预测性:如果一个分子在特定位置断裂,我们可以从整体质量和它碎裂产生的碎片质量的组合中获得更多信息。
知识框 5.15:测量功能相互作用
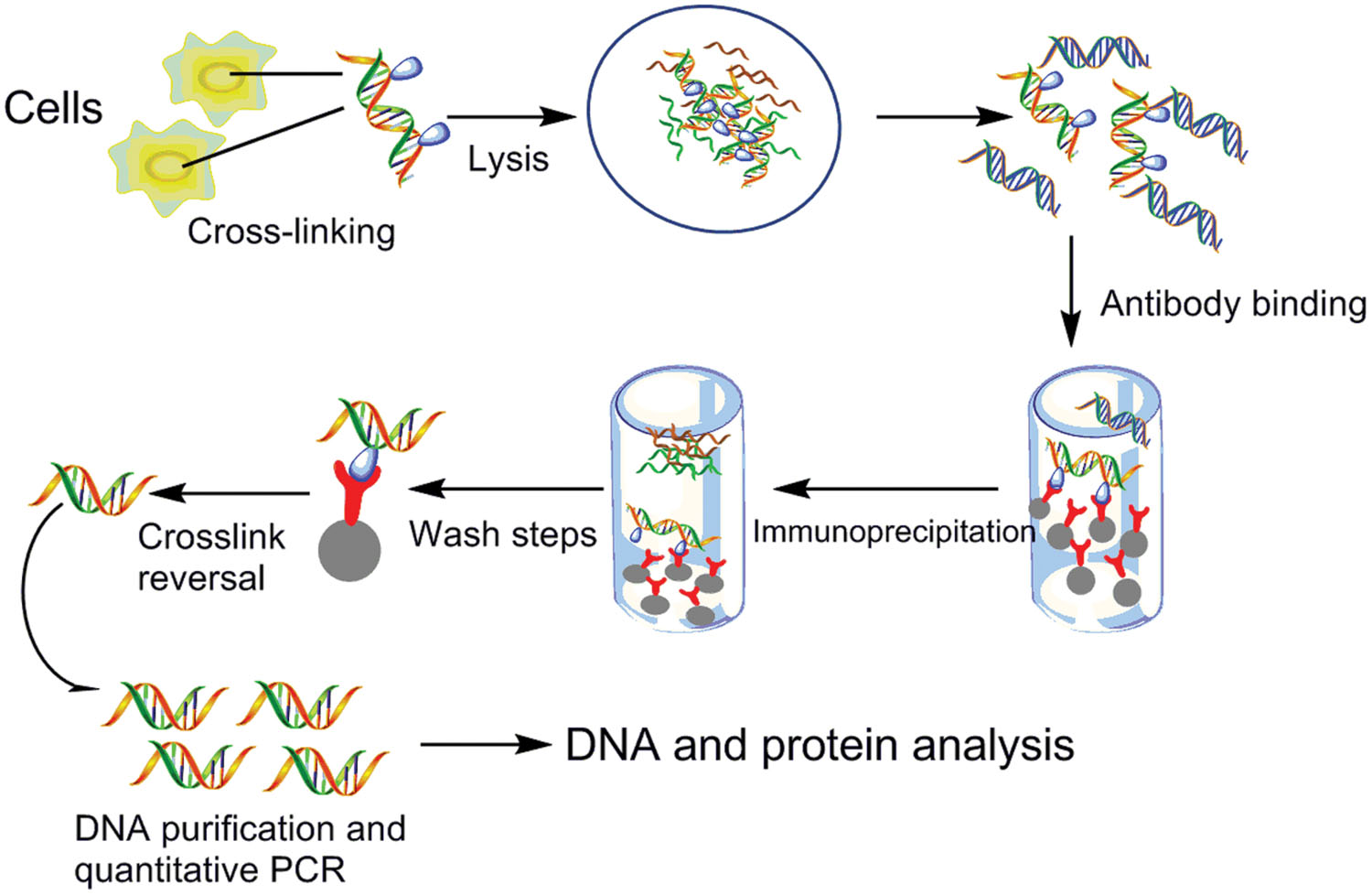
图:染色质免疫沉淀(ChIP)方案。蛋白质与 DNA 交联,之后分离和打碎基因组 DNA。使用抗体,只选择感兴趣的蛋白质(免疫沉淀步骤),之后逆转交联,DNA 可以通过 PCR (ChIP-PCR) 或 NGS (ChIPseq) 测序。当读段映射到基因组时,峰表示蛋白质结合的位置。图片来源:CC BY 4.0 [@chip_protocol_alt_2015]
对于蛋白质-DNA 相互作用,ChIPseq 方法(见上图)使用 RNAseq 来了解蛋白质如何修饰 DNA、启动复制和修复,以及作为转录因子或增强子调控表达。
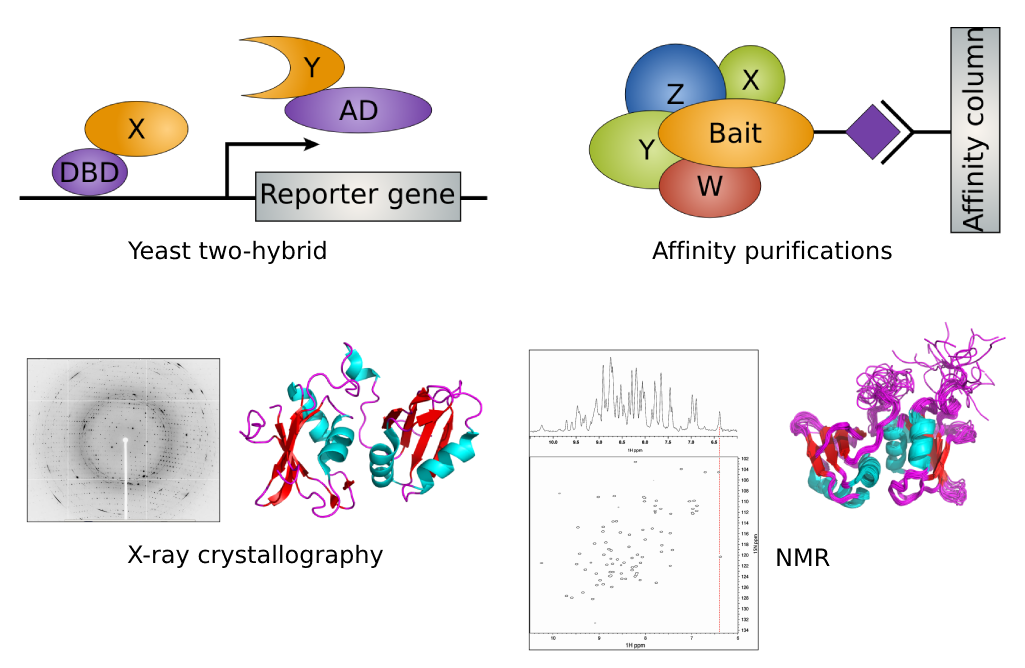
图:检测蛋白质的实验方法。上:高通量,下:低通量。图片来源:CC BY-NC 4.0 上:[@own_5_2024]。下:[@experimental_protein_methods_bottom_alt_nd]
对于蛋白质-蛋白质相互作用,主要的高通量方案(见上图,上)包括:
-
酵母双杂交:将两个蛋白质之一连接到 DNA 结合域,另一个连接到表达激活域。只有当两个蛋白质相互作用时,报告基因(如荧光蛋白基因)才会被表达。
-
串联亲和纯化:与"诱饵"蛋白质相互作用的所有蛋白质被纯化,随后使用 MS 测量。
这些方案有噪声,有较多的假阳性和假阴性,因此通常需要使用低通量方法进行进一步的实验验证——本质上测量蛋白质复合物的结构(见上图,下)。
最近,AlphaFold 3 被引入,承诺在计算上预测蛋白质与其他蛋白质、DNA、小分子等的相互作用(类似于 AlphaFold 2 预测蛋白质结构)。然而,该工具在实践中是否足够可靠仍有待观察;它尚未完全向公众开放,这使得验证不太容易。幸运的是,开源替代方案最近已成为可用,例如 Boltz-2。
参考文献¶
{{ bibliography }}
术语表¶
- 第二代测序 (2nd generation sequencing)
- 见下一代测序
- 分类 (Classification)
- 基于组学数据预测感兴趣的输出(如表型)。
- 聚类 (Clustering)
- 在数据集中寻找组以了解共同因素,即相似的疾病(当聚类样品时)或相似的功能(当聚类基因时)。
- 重叠群 (Contig)
- 基因组组装的一部分,可以无问题或无间隙地组装的连续序列。
- 共分离 (Co-segregation)
- 基因组的哪些部分是从母亲或父亲一起遗传的。
- 缺失 (Deletion)
- 与参考相比,基因组中少量核苷酸(<100bp)的缺失。
- 差异丰度分析 (Differential abundance analysis)
- 比较条件、细胞类型、菌株等之间的水平,通常基于统计检验。
- 草图基因组 (Draft genome)
- 基因组组装的初步结果,到重叠群水平,包含大多数基因区域,但其余部分是碎片化的。
- 富集分析 (Enrichment)
- 识别在一组基因/蛋白质中显著高于偶然预期的生物学过程/功能。
- 表观遗传学 (Epigenetics)
- 不改变基因组序列但影响基因表达的可遗传变化。
- 表观基因组学 (Epigenomics)
- 测量整个基因组的表观遗传状态。
- 流动池 (Flow-cell)
- 测序设备的一部分,DNA 被沉积其上并在此进行实际的测序过程。
- 倍数变化 (Fold change)
- 比较两种条件的相对测量值,通常进行 log2 转换以便于解释和可视化。
- 功能注释 (Functional annotation)
- 使用同源方法确定基因组组装中结构注释基因的假定功能。
- 功能基因组学 (Functional genomics)
- 研究基因/蛋白质功能和相互作用的学科领域。
- 功能蛋白质组学 (Functional proteomics)
- 测量蛋白质与其他蛋白质、DNA 等之间的相互作用。
- 热图 (Heatmap)
- 将数据矩阵作为图像的视觉表示,单元格颜色反映数值。通常沿一个或两个轴进行聚类,以帮助识别具有相似表达水平的样品/基因组。
- 高通量 (High-throughput)
- 收集大量测量数据而无需大量人工干预的技术。
- 杂交 (Hybridisation)
- 两个不同但密切相关物种的个体之间的成功交配,产生了后代。
- 插入 (Insertion)
- 与参考相比,基因组中少量核苷酸(<100bp)的插入。
- 基因渗入 (Introgression)
- 源自不同物种的基因组部分。两个物种在过去某个时间点杂交的结果。
- 比对/映射 (Mapping)
- 在基因组中找到(近似地,允许错误)匹配给定短 DNA 序列(如测序读段)的位置。
- 质谱 (Mass spectrometry, MS)
- 基于电离、分离和检测的分子量测量技术。
- 代谢组学 (Metabolomics)
- 测量细胞中代谢物的存在或水平。
- 微阵列 (Microarrays)
- 基于互补 DNA 序列与附着在表面特定位置的 DNA 结合的荧光来测量基因表达的设备。已被 RNAseq 取代,但在数据库中仍广泛存在。
- 多核苷酸多态性 (MNP)
- 与参考相比,基因组中若干相邻核苷酸的变化。
- 质荷比 (M/z)
- 质谱设备的测量单位:质量与电荷的比值。
- 下一代测序 (Next-generation sequencing)
- NGS,也称第二代测序;Sanger 测序之后的测序技术,主要指 Illumina 测序。
- 组学 (Omics)
- 研究某事物的整体;例如所有基因的表达(转录组学)。
- 双端读段 (Paired-end read)
- 在 Illumina 测序中,来自同一 DNA 片段的两条读段:第一条从一个端测序,第二条从另一端以反向互补测序。当原始 DNA 片段长于两条读段时,比对到参考序列后两条读段之间会有间隙。
- 表型组学 (Phenomics)
- 对组织和生物体的宏观表型(性状)的测量。
- 主成分分析 (Principal Component Analysis, PCA)
- 将具有多个测量的数据投影到保留尽可能多变异的方向上。通常用于投影到 2 维以便可视化分析给定数据集。
- 蛋白质组学 (Proteomics)
- 对细胞中蛋白质的测量。
- 定量蛋白质组学 (Quantitative proteomics)
- 测量细胞中蛋白质的存在/缺失或水平。
- Q 分数 (Q-score)
- 质量分数,用于指示测序中碱基的质量,或在变异检测中表示变异的质量。
- 重复区域 (Repeat region)
- 在基因组中重复多次的 DNA 序列。各拷贝之间可能略有差异。
- RNAseq
- 通过 DNA 测序技术进行的转录组学测量,在将 RNA 转换为 cDNA 之后。
- 鸟枪法蛋白质组学 (Shotgun proteomics)
- 通过 MS 在已知切割位点碎裂成肽段后测量蛋白质水平,然后在蛋白质序列数据库中查找肽段。
- 剪接比对/剪接映射 (Spliced mapping / spliced alignment)
- RNAseq 读段的比对,考虑到内含子在转录本中不存在,因此读段可能部分比对到两个相邻位置。
- 分裂读段 (Split reads)
- 在序列比对过程中被分裂为两部分的读段,两部分比对到参考序列的不同区域。另见剪接比对。
- 结构注释 (Structural annotation)
- 确定基因组组装中(蛋白质编码)基因的位置和特征。
- 系统生物学 (Systems biology)
- 通过构建和迭代更新模型来整体研究复杂生物系统的方法。
- 端粒到端粒组装 (Telomere-to-telomere assembly)
- T2T;覆盖生物体所有序列的基因组组装,无间隙,从一条端粒到另一条端粒组装到每条染色体。
- t 检验 (t-test)
- 广泛使用的统计检验,用于检验两个分布均值之间的差异。它假设数据服从正态分布,但实际中往往并非如此,因此使用更复杂的检验。
- 时间序列分析 (Time series analysis)
- 分析随时间采集的测量值,以研究变化。
- 转录本定量 (Transcript quantification)
- 对 RNAseq 读段计数进行转换和标准化(针对基因长度、文库大小等)以供进一步分析。
- 转录组学 (Transcriptomics)
- 测量细胞中所有基因的表达水平。
- 全基因组鸟枪法测序 (Whole genome shotgun sequencing)
- WGS。同时测序基因组的所有部分,无需预先知道哪部分是哪部分。
本站总访问量 次