第四章
课程来源与翻译声明
- 原著版权:本教程翻译自瓦赫宁根大学(Wageningen University & Research)的经典开源课程 Introduction to Bioinformatics。
- 翻译说明:本版本主要采用人工智能(机器翻译)进行全文本地化,并由团队进行了初步校对。部分专业术语可能存在翻译不够地道或准确的地方。
- 勘误反馈:若你在学习过程中遇到语病、错别字、代码失效或概念歧义,欢迎随时点击页面右上角的 :material-edit: 编辑此页(或联系课题组/在 GitHub Issue 中)提交反馈,帮助我们持续完善本教程。
在本章中,你将学习从氨基酸序列预测蛋白质结构的方法。
学习目标
学习本章后,你应该能够: - 解释蛋白质序列-结构-功能范式 - 讨论预测生物分子结构的重要性 - 列举并解释预测蛋白质二级和三级结构,以及跨膜区和信号肽序列的主要方法 - 应用多种当前方法预测二级结构元件(NetSurfP3.0),包括跨膜区(DeepTMHMM)和信号肽(SignalP),以及三级结构(AlphaFold-PSD、SWISS-Model、Foldseek) - 评估二级和三级结构预测的置信度 - 选择一种(或多种组合)方法来推导氨基酸序列的结构和功能,并分析结果及其质量
蛋白质结构与功能¶
在过去几十年中,大量序列已被获取。然而,对于其中大多数序列,我们尚不知道它们代表什么蛋白质,以及具有什么功能。
蛋白质对地球上的生命至关重要。它们在生物体中具有多种功能,例如支持组织、器官或细胞结构(如我们皮肤中的角蛋白),执行酶促反应(如植物中的 1,5-二磷酸核酮糖羧化酶/加氧酶,又称 Rubisco),或作为介导细胞间通讯的信号转导受体。 正如在第 1 章中所讨论的,仅约 20 种氨基酸构建块构成了所有生命形式中结构极其多样的蛋白质库的基础。如今,氨基酸序列已经大量可获得,要么通过蛋白质组学测量直接获得,要么通过翻译基因组序列间接获得。 在第 1 章中,你还学习了蛋白质是以氨基酸链的形式合成的,由肽键连接,即多肽链(一级结构),然后根据氨基酸侧基之间的各种相互作用折叠成三维(三级)结构。 通常,在这一折叠过程中,较短的局部二维(二级)结构会首先形成,由氢键维系。 有趣的是,尽管蛋白质的氨基酸序列可能不同,但它们的折叠仍然可能产生相似的多肽链三维结构——这些结构具有可比性甚至相似的功能,并在进化时间尺度上被保守(见图:肌红蛋白)。 最后,多个折叠的多肽链可以形成四级复合物。 蛋白质折叠过程非常重要,因为它决定了多肽链的三维结构,而错误折叠可能导致蛋白质功能异常,例如通过与其他蛋白质的非特异性结合导致人类疾病,或在植物中导致功能较差的突变体。总之,要理解蛋白质的功能,了解其三维结构是关键。因此,基于蛋白质序列信息预测蛋白质结构几十年来一直是生物化学家和广大科学界高度关注和具有重大意义的课题。
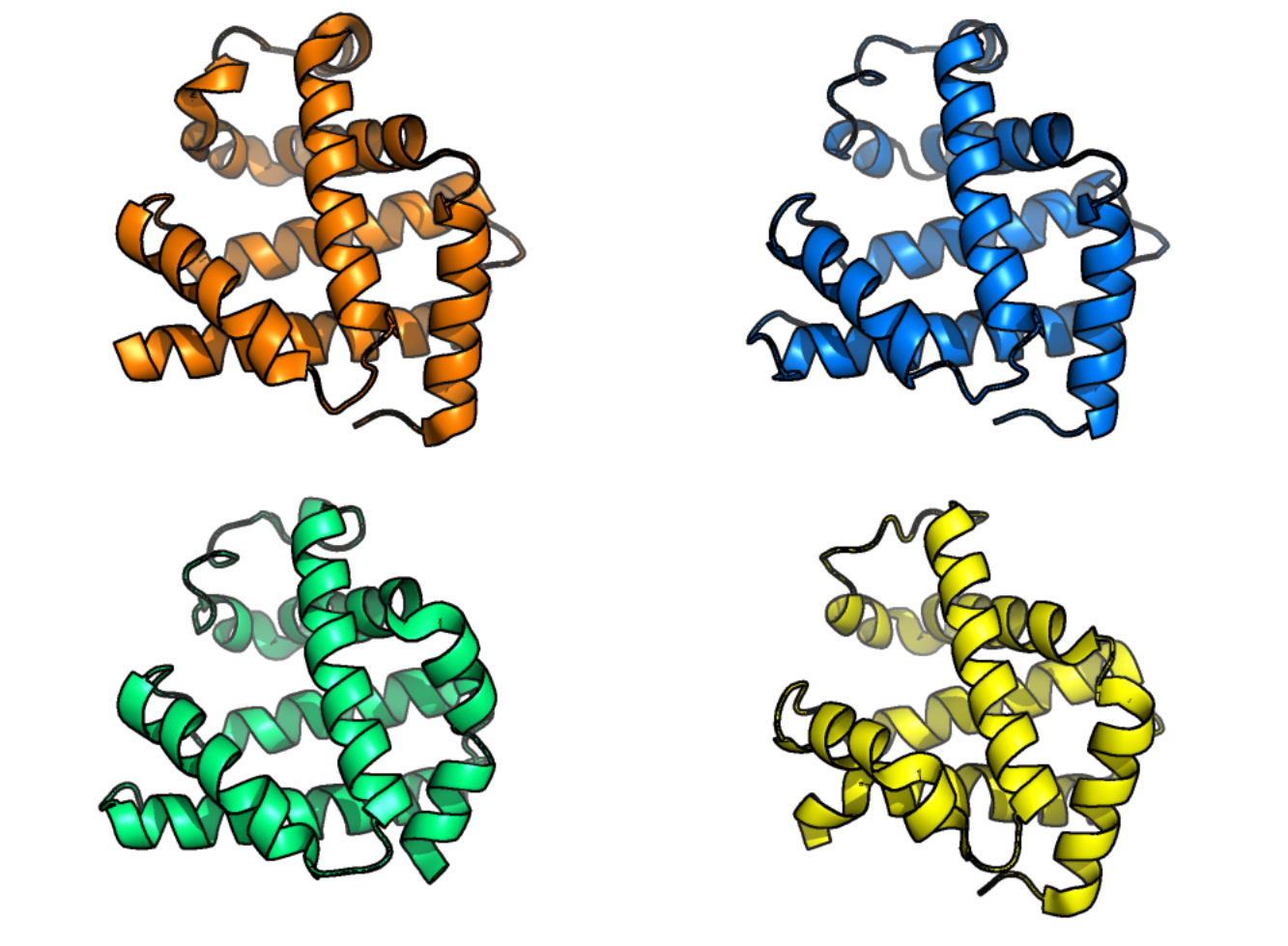
人类肌红蛋白(左上)、非洲象肌红蛋白(右上,与人类蛋白质序列 80% 的序列一致性)、黑鳍金枪鱼肌红蛋白(右下,与人类蛋白质序列 45% 的序列一致性)和鸽子肌红蛋白(左下,与人类蛋白质序列 25% 的序列一致性)的蛋白质结构。肌红蛋白存在于肌肉中,其主要功能是为肌肉细胞供氧。本图中的蛋白质结构说明了即使序列差异较大,结构也可能基本相同。 [@blopig_2021]
实验性蛋白质结构测定¶
虽然当前的生物信息学方法可以从基因组测序的生物体中生成许多假设蛋白质序列,但实验性地测定相应的三维蛋白质结构仍然困难和昂贵。 使用的主要传统实验分析技术是核磁共振(NMR)光谱学和 X 射线晶体学。 前者会产生有用但通常有噪声的测量结果,因为会生成多种结构构象(即组成氨基酸的空间排列,共同形成三维形状);而后者更准确,但也更昂贵。此外,从数据中完全阐明蛋白质三维结构可能需要一年甚至更长时间,而且有些结构根本无法测量,例如由于相关蛋白质的结晶问题。 幸运的是,受技术进步的推动,生物序列数据已变得广泛可获得,主要以基因组序列的形式存在。 通过使用你在第 1 章中学到的遗传密码将这些 DNA 序列翻译为可能的氨基酸序列,可以推断出氨基酸序列并预测理论蛋白质。 然而,不同生物序列的庞大数量使得手动分析这些预测的蛋白质序列变得过于艰巨。因此,需要替代方法来推导三维蛋白质结构,以解读近几十年来可获取的大量生物序列数据。
序列-结构-功能范式¶
序列-结构-功能范式指出,原则上,预测蛋白质折叠及其三维结构,并最终预测其功能的所有信息都存储在其一级序列中。 然而在实践中,从序列预测结构被证明是一项非常复杂和具有挑战性的任务。基于序列预测蛋白质结构和功能比该范式所描述的更为复杂的原因之一,是由于蛋白质局部的二级(二维)结构元件之间既存在短程也存在长程的相互作用。这些相互作用通常形成锚点,三级(三维)结构在此基础上构建。 本章首先描述二维结构指派和预测,然后讨论三维结构预测方法,包括主要挑战和三级结构预测的三个区域。 最后介绍预测和比较三级结构的最新方法:AlphaFold 和 Foldseek。
Attention
序列-结构-功能范式指出,原则上,预测三维结构和功能的所有信息都嵌入在其氨基酸序列中。虽然这是 X 射线晶体学或 NMR 实验性三维结构测定的一种更便宜、更具可扩展性的替代方案,但使用生物信息学方法从序列预测结构被证明是一项非常复杂和具有挑战性的任务。
蛋白质二级结构预测¶
蛋白质由若干局部定义的二级结构元件组成,其中 alpha 螺旋(α-螺旋)和 beta 链/折叠片(β-链/折叠片)是最常见的。 请注意,β-链指的是β-折叠片的一侧,两条链结合在一起形成折叠片结构(正如我们在第 1 章中所学到的)。 在本节中,我们将介绍如何使用三维蛋白质结构来指派这两种二级结构元件,以及如何基于序列数据来预测它们。 此外,我们将介绍两种生物学上重要的二级结构元件"特例"的预测:跨膜区和信号肽。
二级结构指派:标注与准确性¶
如果我们观察折叠蛋白质链的三维结构,通常会发现在结构上更有序的三维结构部分比更随机形状的部分包含更多的α-螺旋和β-链片段。 由于这些二维元件是局部折叠的,它们可以启动更复杂的三级折叠模式的形成。事实上,α-螺旋和β-折叠片的存在/缺失及其特定构象有助于对蛋白质进行分类。 因此,存在根据特定折叠类型以分类的、层次化的方式对蛋白质进行分类的数据库。 因此,将氨基酸残基指派为α-螺旋、β-链或"无规卷曲"(即"其他"),基于蛋白质链的三维结构,可以被视为理解蛋白质结构配置的第一步。 此外,当我们需要训练数据来基于一级(一维)序列预测二级结构元件时,我们需要足够多基于实际结构指派的标注数据。 因此,开发了几种指派工具来替代上述基于已知三维信息手动指派二级结构元件的艰巨任务。
上述氨基酸残基的三种选择将转化为所谓的"三态模型"(α-螺旋、β-链或其他),由 DSSP、PALSSE 和 Stride 等二级结构指派工具使用。这些工具使用三维结构作为输入来分配三种二级结构标签。 在实践作业中,你将获得这些工具结果解释的实践经验。请注意,还有其他——不太常见的——可识别的二级结构元件,例如β-转角(蛋白质链中的急剧弯曲)和几种特殊螺旋。 因此,一些工具将返回八种甚至更多状态。不过,这些也可以归入上述三种原始状态中。
只有当你对来自特定工具的指派/预测的准确性有所了解时,指派或预测蛋白质的二级结构才有用。 任何准确性预测或测量都有助于估计当工具处理未知结构的序列时的性能。 准确性可以基于单个残基的预测来衡量,也可以基于正确预测的螺旋和链的数量来衡量。 一种常用且相对简单的方法是为单个残基分配准确性的 Q3 指标。 Q3 的值可以在 0 到 100% 之间,100% 等同于完美预测。 完全随机预测的值取决于我们预测的不同标签或"状态"的数量:如果我们在"三态模型"中预测,随机预测将返回 33% 的 Q3。 请注意,在研究预测的二级结构指派与正确指派之间的实际比对时,相同的 Q3 分数可能意味着完全不同的情况;因此,需要将数值与实际序列比对一起考虑。 虽然追求完美预测似乎很自然,但通常可达到的最大 Q3 约为 80%。 这主要是由于难以精确定义二级结构元件的起始和结束位置:不同的工具在指派或预测二级结构元件的边界处可能存在偏差。 即使是作为人类,手动标注二级结构元件也可能在哪些残基位于二级结构元件内部或外部方面带来挑战。 在实践作业中,你将进一步探索这一现象。
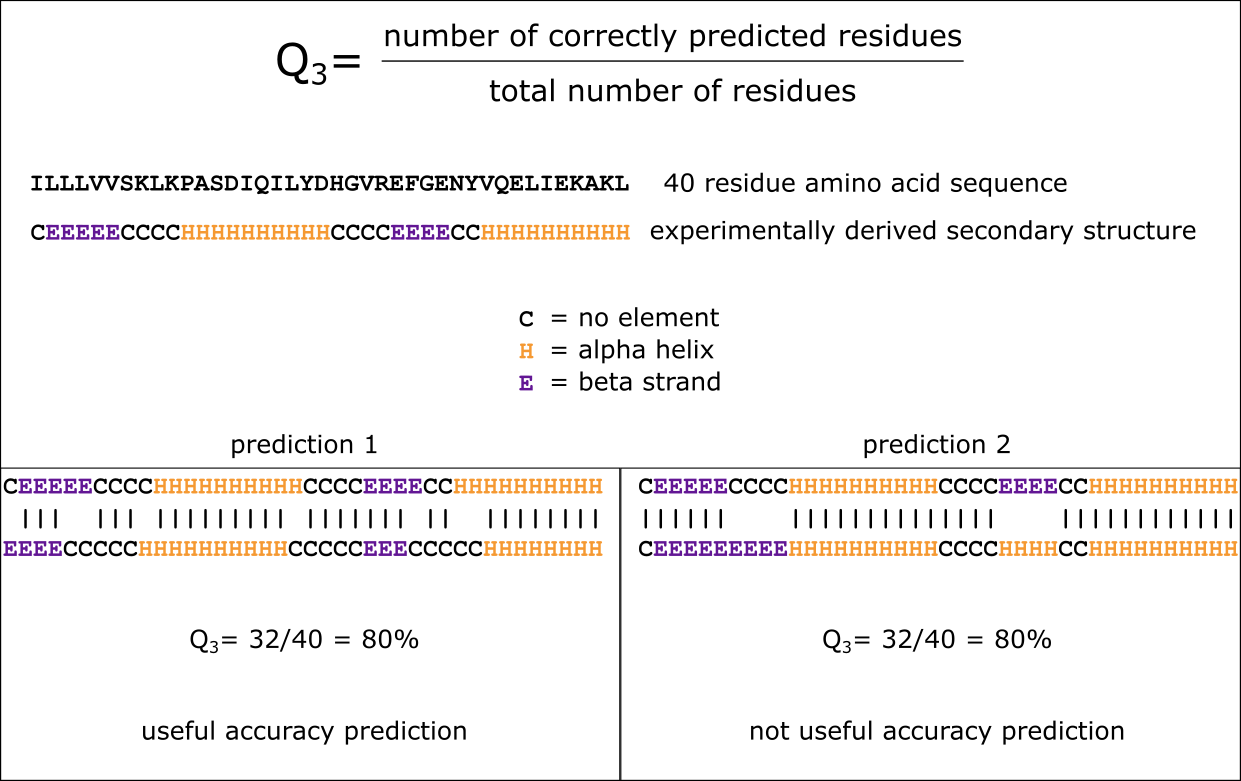
Q3 指标在预测的二级结构结果与实际结构相比存在轻微偏移时(预测 1)能产生有用的准确性预测。然而,当二级结构元件被错误解释时(预测 2 中 E → H),该指标则不再有用。 [@own_4_2024]
二级结构预测:基于统计和机器学习的方法¶
在这里,我们将描述过去几十年来基于序列数据预测二级结构元件的方法。 由于这些方法构成了三级结构预测工具的基础,我们将首先研究它们。
最早的预测二级结构的方法之一是使用统计推断残基的二级结构,即所谓的 Chou-Fasman 方法。自 20 世纪 70 年代以来,该方法使用不断增加的参考蛋白质三维结构和二维结构指派数据来确定每种氨基酸类型形成、破坏或不倾向于形成或破坏α-螺旋或β-链的天然倾向性(见图:Chou-Fasman 方法)。 如果我们考虑一段氨基酸序列,这些倾向性有助于确定α-螺旋或β-链在何处开始或停止。 例如,某些氨基酸有很强的形成α-螺旋的倾向(如丙氨酸)或β-链的倾向(如异亮氨酸),而另一些氨基酸则倾向于破坏这些局部结构。 特别值得注意的是,脯氨酸是两种结构元件的强破坏者。 这可以通过脯氨酸特殊的侧基排列来解释:它的侧基与蛋白质骨架融合了两次,使其在 phi(φ)和 psi(ψ)二面角方面非常不灵活(关于 phi 和 psi 角的更多信息,请参见第 1 章)。 另一种倾向于破坏α-螺旋和β-链的氨基酸是甘氨酸。 虽然 Chou-Fasman 方法已被更精确的统计方法和最近的机器学习方法所取代,但它非常优雅地展示了氨基酸侧基如何影响其形成特定结构的倾向性。
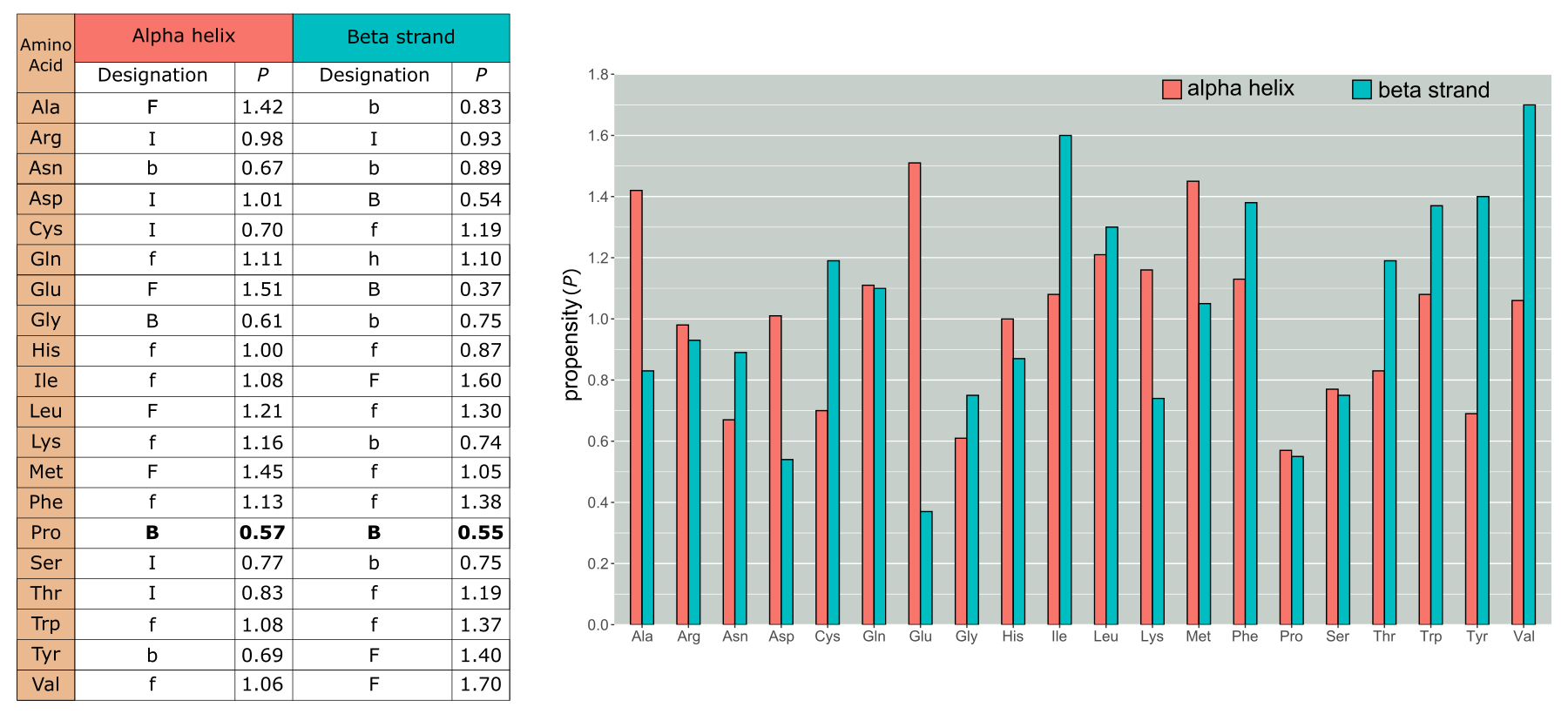
Chou 和 Fasman 倾向性(P)。F 表示强形成者,f 表示弱形成者,B 和 b 分别表示强破坏者和弱破坏者。I(无倾向性)表示既不形成也不破坏螺旋或链的残基。我们可以看到 Pro 具有最低的螺旋形成倾向性,对链的形成倾向性也很低。然而,许多其他弱形成者或无倾向性的残基已经在更多数据可用后被重新分类。 [@own_4_2024]
在随后的几十年中,开发了几种基于统计的方法来改进基于序列的二级结构元件预测。 例如,它们开始纳入多序列比对(MSA——第 2 章中引入并在第 3 章中使用的概念)的信息,包括残基保守性:这些方法首先将查询序列与具有已知三维结构的数据库序列进行匹配,并指派二级结构元件。 然后,使用最佳匹配的序列,通过平均最佳匹配序列的状态来推断查询序列片段中氨基酸残基的二级结构状态,并使用残基保守性等额外信息进行进一步调整。 Zpred 就是这种方法的一个例子。 总的来说,使用多个序列以及关于氨基酸残基理化性质和进化保守性的额外信息极大地提高了预测性能。 在 2000 年代,机器学习以神经网络的形式取代了统计方法。 这种方法使用一个所谓的滑动窗口,包含多个氨基酸残基,其中中心残基的状态使用模型进行预测。 它们通常为每个状态生成概率,可用于分配最可能的状态,即α-螺旋、β-链或无规卷曲(在三态模型中)。 这类方法的例子包括 Jnet 和 RaptorX。
最近,深度学习方法被引入用于基于序列信息预测二级结构元件。 最先进的方法是 NetSurfP,目前已有第 3 版。 在实践作业中,你将获得使用 NetSurfP 3.0 的实践经验。 在这里,我们将简要解释其工作原理以及如何评估其结果。 该预测工具使用深度神经网络方法,同时使用三态和八态定义来准确预测溶剂可及性和二级结构等属性。 为了使这种方法奏效,需要足够多的具有已知 PDB 三维结构的蛋白质链训练数据。 对于某些蛋白质序列,数据库中存在许多近缘变异体,导致某些蛋白质序列片段大量存在。为了避免模型在主要序列片段上过拟合,删除了与测试集中任何其他蛋白质序列具有超过 25% 序列一致性的所有蛋白质序列。 为确保数据质量,选择了分辨率不低于 2.5 埃的结构。 结果产生了约 10,000 条用于训练的蛋白质序列。 为了获得"真实标签",使用 DSSP(见二级结构指派)从相应的蛋白质结构中计算溶剂可及性和二级结构状态等属性,从而生成带有这些属性标签的训练数据集。 神经网络参数使用小批量的蛋白质序列及其"真实标签"进行训练,最终得到一个模型。
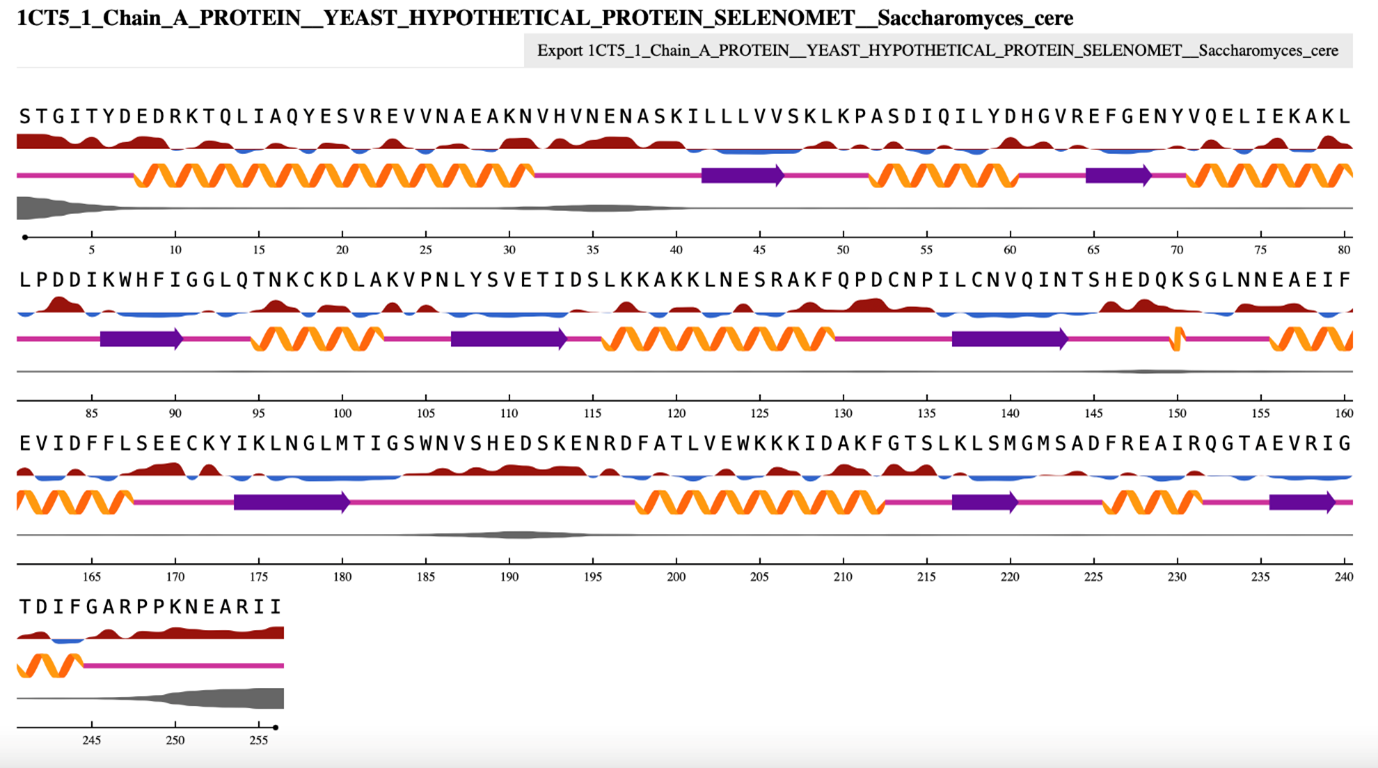
含有α-螺旋和β-链片段的酵母蛋白质的 NetSurfP 3.0 输出。 [@netsurfp_2022, @1CT5_1999]
正如我们在第 1 章中所见,α-螺旋和β-折叠片之间存在关键的相似性和差异性。 这两种二级结构元件都依赖于多肽链中骨架原子之间的氢键。 然而,参与α-螺旋的残基仅具有链内的局部相互作用,而β-折叠片残基可以具有长程相互作用。 因此,对于基于序列的预测工具来说,β-折叠片更难预测。 充足的同源蛋白质的可获得性可以缓解这一瓶颈,并提供可靠的β-折叠片预测。 此外,三维结构预测工具(见蛋白质三级结构预测)的发展预计将进一步提高仅基于序列信息预测二级结构元件和其他逐残基属性(如表面暴露/溶剂可及性)的能力。
Attention
为了从一级序列预测α-螺旋和β-折叠片等二级结构元件,已开发了几种使用两种主要方法的工具:基于统计的方法和基于机器学习的方法。虽然这些工具在过去几十年中的预测精度不断提高,但 80% 的预测精度通常被认为已接近完美,因为二级结构元件的边界通常难以精确定义。
预测跨膜蛋白区域¶
在大多数细胞蛋白质中,具有疏水侧链的氨基酸残基位于蛋白质三维结构内部,从而有效地与细胞内亲水(极性)环境隔离。细胞被一层膜所包围,这层膜将细胞内部与外部世界分隔开来。 因此,如果消息需要从细胞外部传递到内部,或反之,这些消息必须穿过膜。细胞膜由脂质双层组成,其亲水头部朝向外部和细胞环境,疏水酰基链彼此相对。 为此,细胞机制利用蛋白质来协助信号跨膜转导。 球状蛋白存在于细胞内,而所谓的跨膜蛋白至少跨越膜一次。 据估计,真核细胞中约 30% 的蛋白质是跨膜蛋白,表明其功能重要性。 由于膜在大小(脂质双层的厚度)和极性(亲水外侧和疏水内侧)方面的特殊环境,通常只有某些局部结构构型能够跨越膜。 这些构型通常与蛋白质的功能相关:无论是信号转导的受体还是跨膜运输特定物质的转运蛋白。
首先让我们考虑大小:膜的平均厚度约为 30 埃(Å),这对应于 15 到 30 个残基的α-螺旋才能适应膜层。
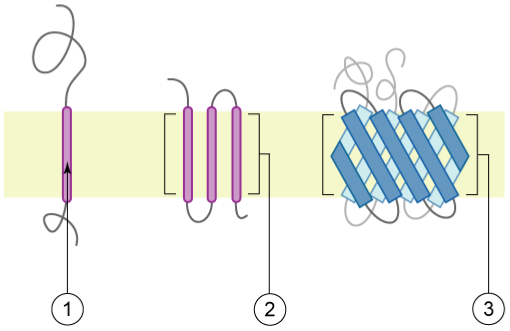
含有跨膜区的蛋白质(又称跨膜蛋白)的示意图,膜以浅黄色表示:1)含有α-螺旋的单次跨膜蛋白(单通道)2)含有α-螺旋的多次跨膜蛋白(多通道)3)含有β-折叠片的多次跨膜蛋白。 [@transmembrane_proteins_2006]
最简单的局部跨膜元件是一个约 15-30 个氨基酸残基的α-螺旋,主要由非极性侧基组成(图:跨膜蛋白,1)。 其长度受脂质双层厚度的限制,而非极性侧基将与膜中脂质的酰基链产生有利的相互作用。 跨膜蛋白可以跨越膜一次或多次(即图:跨膜蛋白,1 和 2),在某些情况下,跨膜蛋白的特殊构型会创建一个"孔道"状结构。 在这里,某些螺旋残基可以带电荷,因为孔道环境完全与膜双层隔离。 另一种常用的跨膜构型是"β-桶"。该元件由 8-22 条跨膜β-链组成(尽管可能存在具有更多β-链的更大结构),共同形成"桶状"(图:跨膜蛋白,3),有效地将桶内部与外部隔开,从而在膜中创建一个孔道。 此类孔道可以通过蛋白质片段的"开关"形式来密封,该片段处于"开放"或"关闭"构型。
信号肽¶
蛋白质在细胞中的合成地点通常不是其发挥作用的地方。 为了在正确的位置发挥功能,蛋白质需要从其折叠和形成的地方运输出去。 细胞机制开发了一个使用"肽标签"的信号系统,以实现多肽链向其作用位点的有效运输。 这些"标签"被称为信号肽。它们是细胞转运机制识别的肽信号。 存在信号肽的典型作用位点包括细胞膜和内质网。 此外,信号肽可以将蛋白质引导至从溶酶体分泌或导入。 通常,在蛋白质到达目标位置后,信号肽序列会被酶促地从蛋白质上去除。 因此,信号肽的存在可以根据氨基酸序列为蛋白质的作用位点提供重要线索。
信号肽是位于蛋白质序列 N 末端的引导氨基酸序列,由约 15-30 个残基组成(图:信号肽)。 细胞转运机制对信号肽的实际识别并非基于保守的氨基酸序列,而是很大程度上取决于信号肽中氨基酸的理化性质。 信号肽通常由三个区域组成:第一个区域(n 区)通常含有 1-5 个带正电荷的氨基酸,第二个区域(h 区)由 5-15 个疏水氨基酸组成,第三个区域(c 区)含有 3-7 个极性但大多不带电荷的氨基酸。
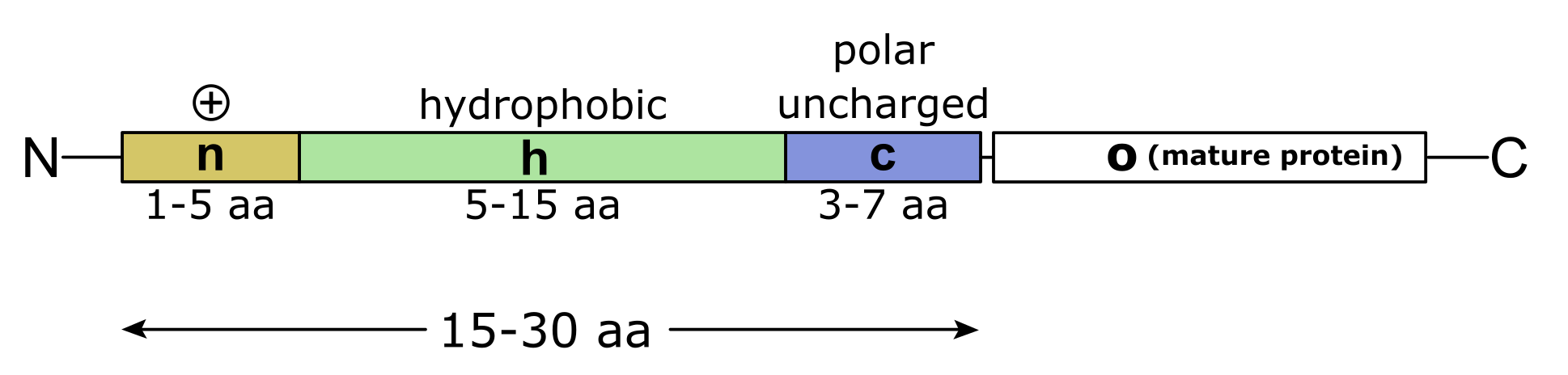
信号肽示意图及其带正电荷的 N 末端、疏水核心区(h 区)、极性(大多)不带电荷的 c 区和成熟蛋白质(o)。 [@signal_peptide_2010]
基于序列的跨膜区和信号肽预测¶
鉴于其功能线索,仅基于氨基酸序列准确预测跨膜区和信号肽在研究未知蛋白质的可能功能时非常有利。 DeepTMHMM 目前是预测蛋白质序列中跨膜区和信号肽的最优秀的工具。 该程序为序列中的每个氨基酸预测几个标签:信号肽(S)、细胞内/细胞质(I)、α-螺旋膜蛋白(M)、β-桶膜蛋白(B)、周质空间(P)和细胞外/内质网/高尔基体/溶酶体腔(O)。
跨膜区和信号肽在很大程度上由构成它们的氨基酸残基的理化性质所定义,而不是由保守的基序或短残基序列所定义。 这使得使用基于比对的经典方法识别这些二级结构元件变得非常困难。 然而,使用机器学习方法,可以学习已知序列训练数据集的特征并用于未知数据的预测。 训练后的模型可以判断未知序列中氨基酸的性质,从而允许识别跨膜区和信号肽。 因此,DeepTMHMM 使用一个深度学习模型,将蛋白质序列作为输入,然后输出相应的逐残基标签。 综合所有信息并考虑其在氨基酸序列中的顺序,残基标签定义了预测的蛋白质拓扑结构。 值得注意的是,DeepTMHMM 和下文解释的 SignalP 都返回概率(另见第 3 章中概念上类似的似然值),而不是绝对预测,这在直接提示预测可靠性方面是有利的。 DeepTMHMM 可以预测五种不同的拓扑结构,即无信号肽的α-螺旋跨膜蛋白(alpha TM)、有信号肽的α-螺旋跨膜蛋白(SP + alpha TM)、β-桶跨膜蛋白(Beta)、有信号肽的球状蛋白(SP + Globular)和无信号肽的球状蛋白(Globular)。 重要的是,这里预测的两种二级结构元件具有共同性质,深度学习模型需要足够的示例数据来区分跨膜区和信号肽。
在下图中,你可以观察 DeepTMHMM - alpha TM 对多次跨膜蛋白的典型输出。
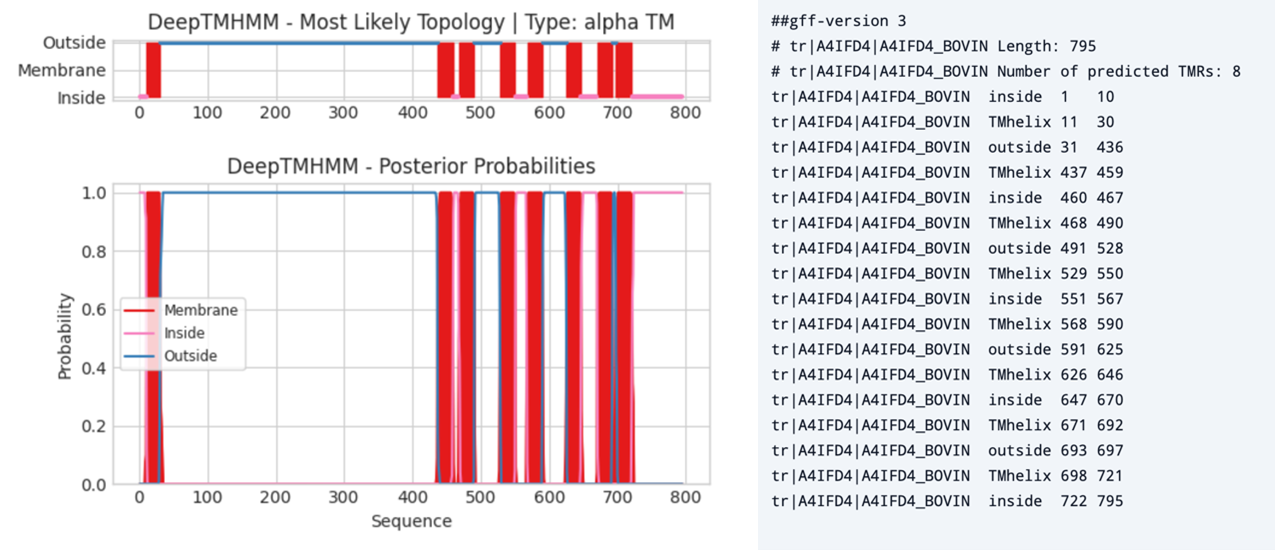
左图:DeepTMHMM - alpha TM 对牛粘附 G 蛋白偶联受体 G7(ADGRG7,A4IFD4)的预测输出。右图:同一蛋白质的 gff 文件,列出了跨膜结构(α-螺旋)的数量、氨基酸位置,以及残基位于膜内还是膜外。 [@deeptmhmm_2022]
下图展示了 DeepTMHMM - beta 对β-桶跨膜蛋白的示例输出。

左图:DeepTMHMM - beta 对鼠伤寒沙门氏菌(Salmonella typhimurium)外膜蛋白 C(前体)(OMPC-SALTY,P0A263)的预测输出。右图:同一蛋白质的 gff 文件,列出了信号肽位置、跨膜结构(β-折叠片)的数量、氨基酸位置,以及残基位于周质空间还是膜外。 [@deeptmhmm_2022]
SignalP¶
上一节介绍了 DeepTMHMM 如何用于预测信号肽的存在;然而,在区分不同信号肽类型方面存在更专门的工具,例如 SignalP 6.0。 该工具可以从序列数据中预测古菌、真核生物和细菌中所有已知类型信号肽的信号肽。 此外,SignalP 6.0 预测信号肽的区域。根据类型,预测 n、h 和 c 区的位置以及其他特征。
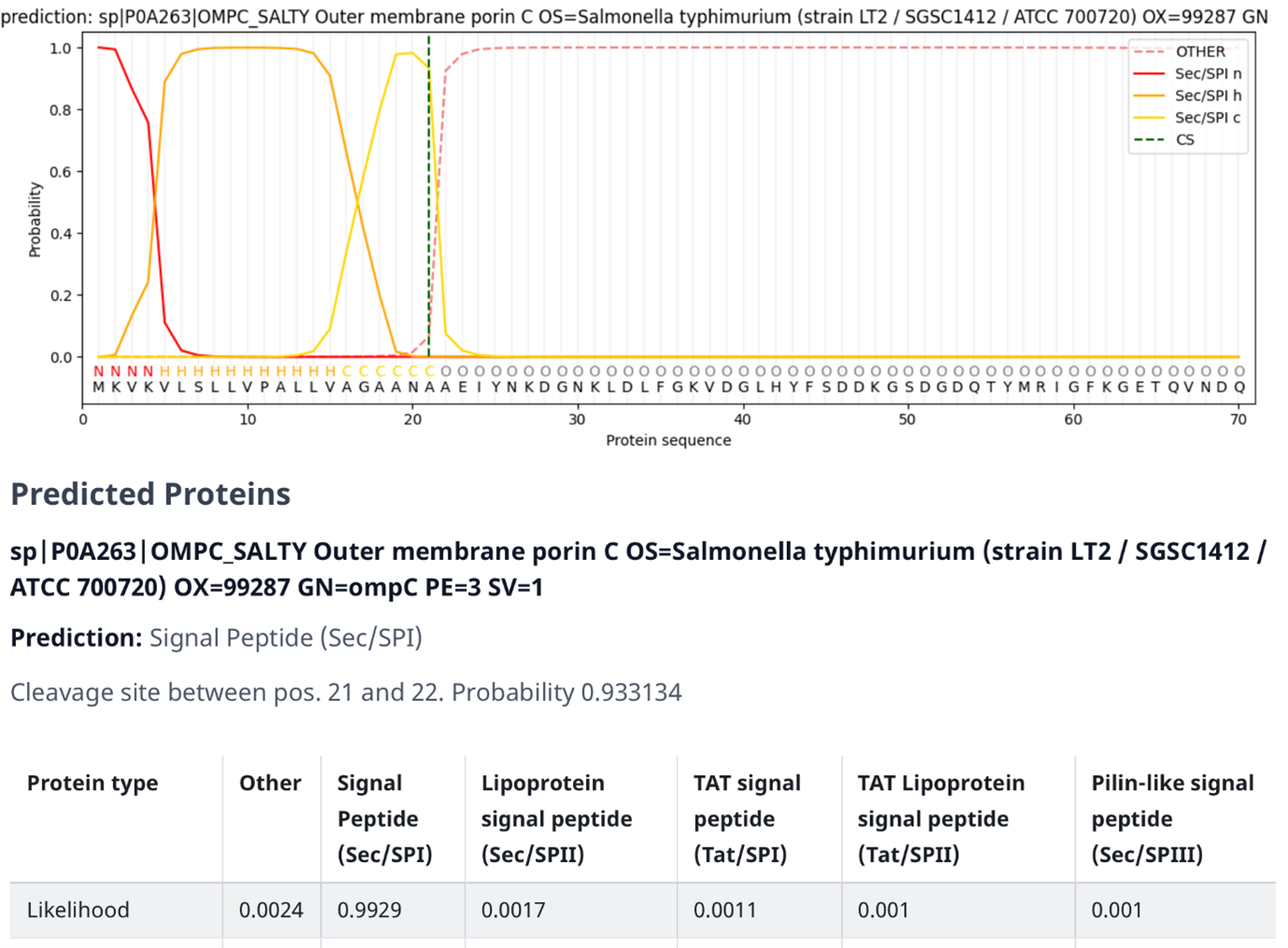
SignalP 对鼠伤寒沙门氏菌(Salmonella typhimurium)外膜蛋白 C(前体)(OMPC-SALTY,P0A263)的输出。 [@signalp_2022]
下图展示了 SignalP 6.0 的示例输出(注意信号肽结构与前面信号肽示意图的相似性)。 该图包含几个元素:
顶部图表由以下元素组成: - 深橙色线(Sec/SPI n)表示特定区域被识别为 N 末端的概率,该概率也在线下方显示为字母 "N"。 - 浅橙色线(Sec/SPI h)表示特定区域被识别为 h 区的概率,该概率也在线下方显示为字母 "H"。 - 黄色线(Sec/SPI c)表示特定区域被识别为 c 区的概率,该概率也在线下方显示为字母 "C"。 - 虚线橙色线(OTHER)表示特定区域被识别为信号肽子部分以外的其他内容的概率,例如成熟蛋白质本身。 该概率也在线下方显示为字母 "O"。 - 虚线绿色线(CS)表示特定区域被识别为切割位点的概率,即信号肽与蛋白质序列其余部分分离的位置。 - 字母下方的蛋白质序列,指示某个区域属于哪个部分。
信号肽评分(橙色线)是通过对信号肽和其他序列的区分进行训练的,如果相应氨基酸是信号肽的一部分,则该值较高。 因此,成熟蛋白质的氨基酸具有较低的信号肽评分。 最大切割评分(虚线绿色线)出现在成熟蛋白质第一个氨基酸的位置,即切割位点之后一个位置。 切割评分分析是通过对信号肽和蛋白质序列之间切割位点的识别进行训练的。
标准分泌信号肽称为 Sec/SPI,由 Sec 转位子转运并由信号肽酶 I(Lep)切割。 还有四种其他信号肽类型(另见下文方框),但它们超出了本课程的范围。 然而,重要的是要知道 SignalP 等工具能够区分不同的信号肽类型,并基于概率对其概率做出准确预测。
下图图表下方信息包含以下元素: - 预测指示给定序列最可能的信号肽类型。 - 切割位点显示蛋白质序列中切割位点的氨基酸位置及其概率。
页面底部的表格包含以下元素: - 不同类型信号肽的似然/概率评分以及完全不是信号肽的概率(Other)。
Attention
跨膜区和信号肽在很大程度上由构成它们的氨基酸残基的理化性质所定义,而不是由保守的基序或短残基序列所定义。标记训练数据量的增加使得能够训练隐马尔可夫模型来从一级序列预测跨膜区和信号肽序列的存在。
另请参阅
在古菌、真核生物和细菌中,SignalP 6.0 可以区分五种类型的信号肽: - Sec/SPI:由 Sec 转位子转运并由信号肽酶 I(Lep)切割的"标准"分泌信号肽。 - Sec/SPII:由 Sec 转位子转运并由信号肽酶 II(Lsp)切割的脂蛋白信号肽。 - Tat/SPI:由 Tat 转位子转运并由信号肽酶 I(Lep)切割的 Tat 信号肽。 - Tat/SPII:由 Tat 转位子转运并由信号肽酶 II(Lsp)切割的 Tat 脂蛋白信号肽。 - Sec/SPIII:由 Sec 转位子转运并由信号肽酶 III(PilD/PibD)切割的菌毛蛋白及菌毛蛋白样信号肽。
关于 SignalP 6.0 中评分的更多信息: Y-score(组合切割位点评分)是切割位点评分绝对值和信号肽评分梯度的几何平均值,显示切割位点评分较高且信号肽评分拐点所在的位置。 此外,还计算了另外两个值。S-mean 是信号肽中所有氨基酸的信号肽评分的平均值。 因此,如果存在信号肽,该值应该较高。 D-score 是 S-mean 值和 Y-score 最大值的算术平均值。 如果预测了信号肽,该值也会较高。
蛋白质三级结构预测¶
首先,需要认识到二级结构元件的预测已经构成了预测蛋白质三维结构工具的基础。 我们将首先探讨三种传统的结构预测方法,然后介绍三维结构预测中最突出的新方法(AlphaFold),它依赖于传统方法的几个概念。
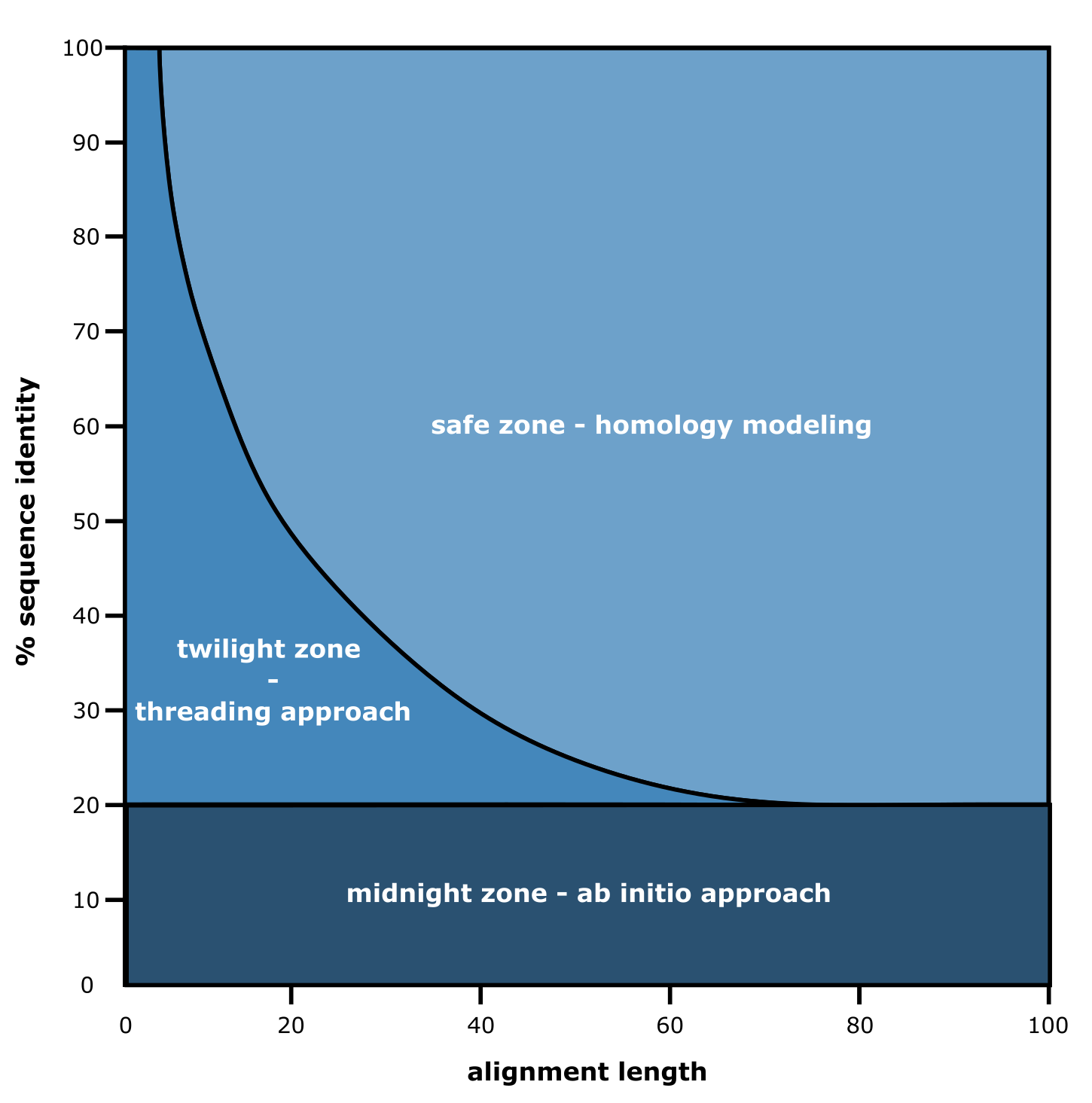
三级结构预测方法的三个区域。 [@own_4_2024]
已提出并使用了多种方法,包括从头计算(ab initio)、穿线法(又称基于片段的建模)和同源建模,以实现从序列到结构的预测,其中序列一致性和比对长度是决定选择哪种方法的最重要因素。 为了在这三种传统结构预测方法之间做出有效选择,提出了所谓的三个区域概念。 根据图表观察,当查询蛋白质序列与实验获得结构的序列之间的序列一致性低于 20% 时,需要使用从头计算方法,字面意思即:从头开始。 由于这种方法计算量大且需要大量专家知识,因此并未被广泛使用。 本质上,这类方法旨在使用氨基酸残基及其环境的理化性质来模拟蛋白质序列的折叠过程。 随着使用的序列长度增加,整个三维结构可能出现越来越多的可能折叠,使其成为一项计算密集型任务。 例如,考虑 100 个氨基酸残基,每个残基都有 psi、phi 和 omega 角。如果它们(仅)每个角有 3 种可能性,这将导致该序列有 3300(= 10143)种可能的折叠。 如果每种折叠只需要 1 秒来评估其现实性和能量有利性,我们将需要 10126 年来分析并提出一个建议的三维结构,而这只是在严重约束条件下的 100 个氨基酸。
幸运的是,实验获得的蛋白质三维结构数据库在不断增长。 因此,你的查询序列有很好的机会具有 >20% 的序列一致性。 如上图所示,序列比对的长度是另一个关键因素:如果匹配的是较短片段,可以使用穿线法(或基于片段的)方法。 这种方法侧重于将这些片段与已知折叠进行匹配,即通常由二级结构元件组成的局部结构。 如果匹配到功能域,这已经有助于推测蛋白质的功能。 由于这种方法也相对计算密集,而且下面讨论的较新方法(即AlphaFold)在识别此类折叠方面更为出色,我们在本课程中不会获得穿线法的实践经验。
如果查询序列一致性和比对长度都足够大,可以尝试同源建模来创建结构模型。 需要在蛋白质结构数据库中找到所谓的"模板序列",使其"足够相似"以作为三维预测的结构蓝图。
如今,SWISS-MODEL 提供预计算的三维同源模型。需要注意的是,SWISS-MODEL 现在也包含基于 AlphaFold 深度学习的模型(见AlphaFold部分)。使用模型的一个关键方面是评估其可靠性。由于同源建模和 AlphaFold 的模型有不同的可靠性检查方式,因此注意三维结构模型的来源很重要。在实践作业中,你将学习如何评估同源建模和 AlphaFold 模型的可靠性。
仍然,几乎每天都有具有与现有蛋白质三维结构相似性很小的新蛋白质序列被发现,从而落入上图中的"午夜区"。 因此,科学界一直在采用各种基于人工智能的方法,其中 AlphaFold 是迄今为止最突出的一个。
Attention
使用与数据库中已知蛋白质序列相似性极低的氨基酸序列预测蛋白质结构仍然是最困难的任务。蛋白质序列与数据库条目的序列相似性越高,就越能从中推断出结构和功能属性。
AlphaFold¶
2018 年,Google 的 DeepMind 团队推出了一种基于机器学习的方法——AlphaFold,随后 2020 年推出了 AlphaFold 2,最新版本 3 于 2024 年发布。在本教材中,我们将主要讨论 AlphaFold 2,并将其简称为 AlphaFold。 本教材描述了 AlphaFold 如何在先前方法的基础上构建,并已经在生物化学和生物信息学领域产生了重大影响。 就其带来的可能性和颠覆性而言,可以将其类比为智能手机的引入:以前,需要去图书馆找电脑连接互联网来获取天气预报,现在只需拿出手机查看天气。 本节解释了为什么 AlphaFold 只能在当前时代开发并发挥作用,如何以公平的方式与其他方法进行比较,它如何依赖数据库搜索和多序列比对,以及包含 AlphaFold 预测结构模型的 AlphaFold 蛋白质结构数据库(AlphaFold DB)的引入对发现流程的意义。
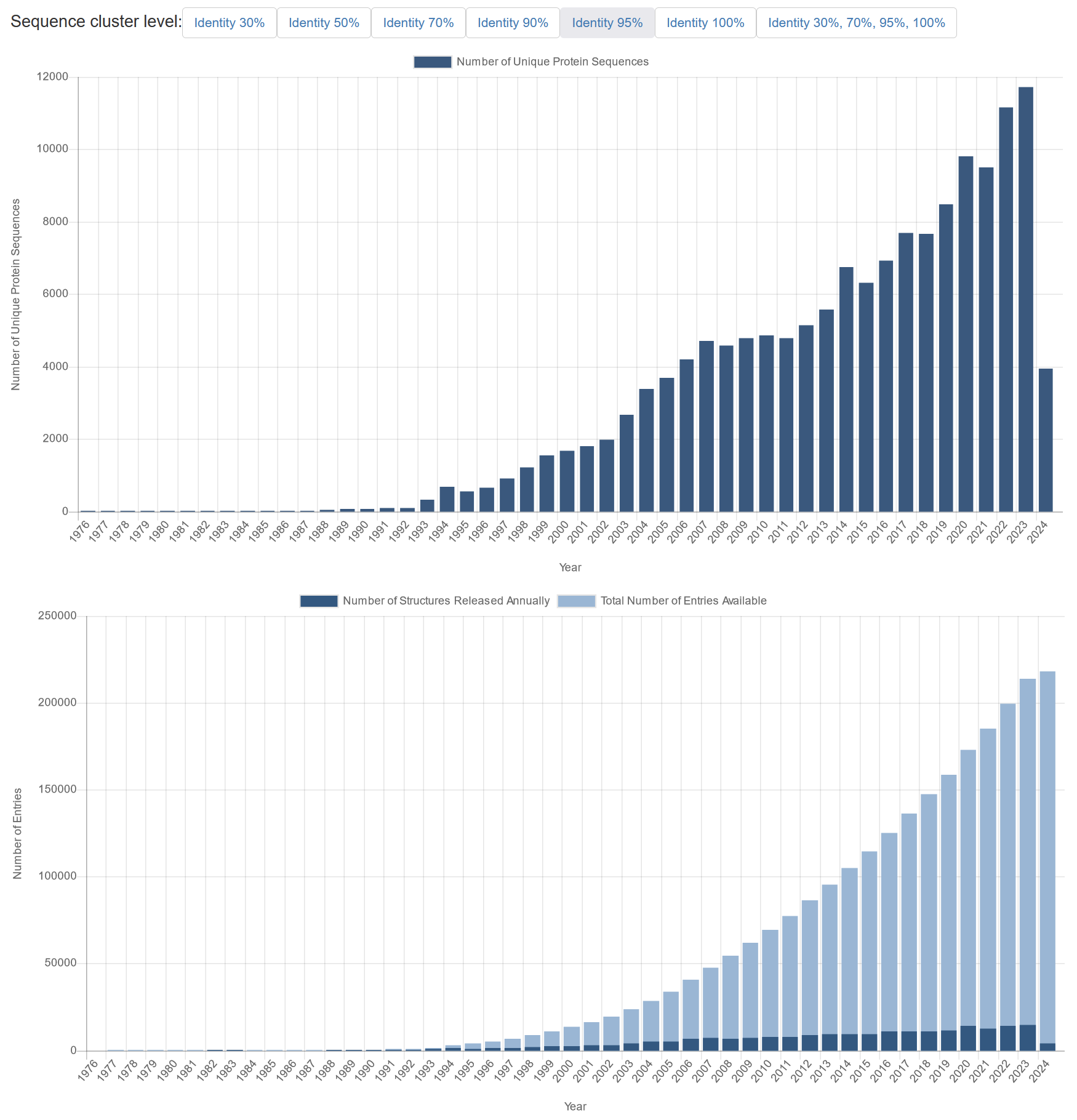
上图:1976 年至 2024 年初每年添加到蛋白质数据库中的独特三维蛋白质结构数量(基于 95% 序列一致性)。下图:1976 年至 2024 年初添加到蛋白质数据库中的实际(深蓝色)和累计(浅蓝色)蛋白质条目总数(即不按唯一性分组)。注意:2024 年结构数量较低是因为这些统计数据是在 2024 年初从 PDB 获取的。 [@PDB_stats_2024]
AlphaFold 方法基于机器学习,即基于训练数据拟合预测模型的计算机算法。 然后,当给定一个前所未见的序列时,该模型可以预测结构。 DeepMind 使用了非常大的神经网络模型,即所谓的深度学习。 此类模型的训练数据理想情况下应包含大量已知示例,以应对蛋白质结构预测等非常复杂的问题。 蛋白质数据库(PDB)收集此类实验训练数据,即测量的蛋白质三维结构。 目前,约 218,000 个 PDB 条目涵盖了约 150,000 个独特蛋白质序列(基于 95% 序列相似性)(上图,上方)。 后一个数字很重要,因为足够多样化的示例集将确保训练数据中有足够的示例来识别各种蛋白质折叠和其他结构特征的相关模式。
作为其最新机器学习模型的输入,DeepMind 团队预测了许多蛋白质序列的结构,在过滤高质量和可靠预测后,将 100,000 条蛋白质序列添加到训练数据中,这种技术称为数据增强。 因此,在模型训练时,团队可以使用约 300,000 个蛋白质序列-三维结构组合来训练其 AlphaFold 模型,该模型以 FASTA 文件作为输入,输出三维结构模型(在评估蛋白质结构模型质量部分描述)。
AlphaFold 对生物化学领域的影响¶
如果没有独立测试数据集,AlphaFold 的真正影响将难以评估。 由于蛋白质折叠和三维结构预测是生物化学的重大挑战之一,蛋白质结构预测关键评估(CASP)竞赛于 1994 年创立。 CASP 是一个全社区范围的竞赛,要求研究小组预测没有任何公开三维结构的蛋白质序列的三维结构。 全球超过 100 个研究小组每两年参加一次 CASP 竞赛。 使用当前可用的所有序列和结构数据,他们预测具有新获得(尚未发布)结构的蛋白质序列的结构,这些结构专门为此竞赛而对外保密。
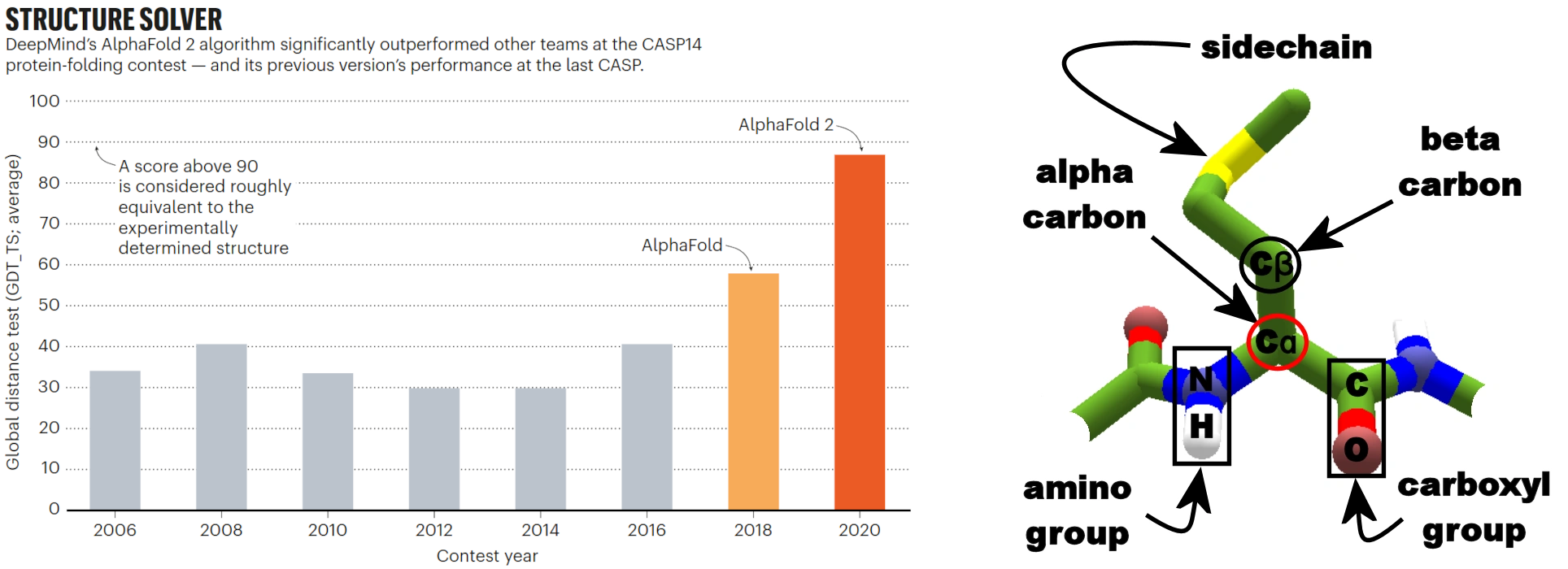
左图:竞赛获胜者的平均 GDT 测试分数。右图:蛋白质骨架中氨基酸残基示意图,标注了关键原子,包括用于 GDT-TS 的α-碳。 [@GDT_2020, @alpha_carbon_2018]
上图左图的 y 轴显示了 CASP 使用的主要评估指标:全局距离测试-总分(GDT-TS),作为所有挑战(即没有公开已知三维结构的蛋白质序列)的平均值。 它测量的是预测结构中氨基酸的α-碳(上图,右图)在最佳可能比对下,与已知结构的阈值距离(准确地说是四个阈值的平均值:1、2、4、8 Å)内的百分比。 上图显示了在多年停滞之后,2018 年 AlphaFold 明显比早年结果有了大幅改进,从而展示了 AlphaFold 对该领域的总体影响。此外,后续 AlphaFold 模型的进展也在 GDT-TS 的增长中可见。 2020 年,他们更新系统 AlphaFold 2 的预测结果如此准确,以至于结构预测问题被一些人称为"已解决"。 2024 年 5 月,Google DeepMind 团队发布了 AlphaFold 3。 虽然下一届 CASP(CASP16)的结果在撰写本文时已经知晓,但尚无 AlphaFold 3 与其前身及其他方法的比较图表。预计 AlphaFold 3 的结果仍然占据主导地位,同时新的挑战包括蛋白质-配体对接,AlphaFold 3 也已开始预测。 需要注意的是,任何预测方法都无法达到 100 分,因为蛋白质结构中存在本质上难以建模的区域,即非常灵活的部分或如螺旋和无规卷曲之间的过渡区域。 因此,90-95% 的分数被认为与实验获得的三维结构同等优秀,AlphaFold 2 几乎达到了这一分数。
综合以上所述,让我们再看一下上图。 2020 年的最大平均 GDT-TS 分数比 2018 年以前的版本翻了一番多。 这意味着对于更多的蛋白质序列,我们可以获得关于其三维结构的某种可靠洞见。 根据序列-结构-功能范式,这也为我们提供了关于其可能功能的洞见。 由于仍有许多功能未知的蛋白质序列,像 AlphaFold 这样的预测软件可以在理解它们在生物化学中的功能和作用方面发挥非常重要的作用。 现在我们更好地理解了 AlphaFold 的影响,让我们进一步了解它的工作原理。
另请参阅
虽然 AlphaFold 3 目前尚无独立的准确性测试结果,但 Google DeepMind 团队在AlphaFold 3 的论文中提供了准确性指标。 他们显示了在单体结构预测方面,AlphaFold-Multimer 2.3 到 AlphaFold 3 之间的边际增长(平均 LDDT 从 85.5 到 86.9),蛋白质-蛋白质相互作用预测方面的显著增长(dockq > 0.23 从 67.5% 到 76.6%),以及蛋白质-抗体相互作用预测方面的非常显著增长(dockq > 0.23 从 29.6% 到 62.9%)。
AlphaFold 的工作原理¶
要使用 AlphaFold 进行预测,你只需要一个包含目标蛋白质一级序列的 FASTA 文件。 AlphaFold 工作的核心是一个复杂的机器学习模型。 然而,它并非从头构建:它大量建立在先前开发的方法之上,以创建可靠的结构模型。 最新的 AlphaFold 实现可以概括为三个关键模块,这些模块与教材中先前的概念和知识相链接。下面将解释这三个模块。
第一个模块将蛋白质序列处理成所谓的数值"表示",可以用作机器学习模型的输入。 为了创建这些表示,首先执行数据库搜索(第 2 章),根据相似性找到最合适的序列。 接下来,创建两种表示(即下图中的上路径和下路径):多序列比对(MSA——第 2 章中引入并在第 3 章中使用的概念),用于捕获序列变异;以及一个表示所有残基之间如何可能相互作用(即在三维结构中彼此接近)的表示,以接触图的形式。 数据库搜索还用于查找 PDB 数据库中是否有合适的"模板"。 最多可以选择四个顶级模板作为预测模型的起始位置。 请注意,这也是同源建模的第一步,而且 AlphaFold 可以仅基于高质量的多序列比对(MSA——见第 2 章)做出"良好"预测;因此,不需要模板。 重要的是要认识到 AlphaFold 很大程度上基于共进化信息。 让我们简要回顾为什么这对结构预测很重要。 你可能已经意识到,残基在蛋白质一级序列中的位置并不反映其在三维空间中的最终位置:一级序列中距离较远的残基在折叠后可能最终彼此靠近,并且可能具有特定的相互作用来稳定三维结构。 共进化的概念意味着如果两个相互作用的残基对蛋白质功能很重要,它们很可能共同进化。 换句话说,如果其中一个变成了不同的氨基酸,另一个也很可能需要改变以维持支持蛋白质三维结构的相互作用。 这种基因组信号只有在我们将许多蛋白质序列相互比较时才能提取。 因此,高质量的深度 MSA 对良好预测至关重要。 在这里,AlphaFold 使用 MSA 来提取进化信号并预测残基的共进化。
第二个模块使用第一个模块的表示,旨在找到蛋白质序列如何折叠成其三维结构的约束。 这部分是实际的机器学习模型,我们将主要将其视为一个黑箱。 该模型使用深度学习来学习哪些输入特征对基于数据驱动模式识别的蛋白质折叠预测是重要的。 模型在序列-残基(MSA)和残基-残基(接触图)表示之间来回传递信息。 这部分需要大量计算时间和精力,因此需要良好的基础设施,而这并非所有实验室都具备。 DeepMind 团队拥有训练大型机器学习模型所需的强大资源。
第三个也是最后一个模块是结构构建器,实际的折叠和结构模型优化在此进行,使用氨基酸原子键的 phi、psi 和 omega 角(另见第 1 章)。 此外,确定局部和全局置信度评分。 通常会进行多次预测循环,预测的三维结构模型作为新的输入(即模板)提供给结构预测,以允许进一步微调。 结构构建器接收来自多个独立训练模型的输入。 这产生多个具有微小或较大差异的三维结构模型,最终根据模型的置信度评分进行排名(见评估蛋白质结构模型质量)。
总结 AlphaFold 的过程:进行数据库搜索以构建 MSA 和查找模板,将完全相同的输入提供给多个具有略微不同参数设置的相同机器学习模型,结构构建器为它们创建三维结构模型,并根据置信度评分进行排名以报告表现最佳的模型。
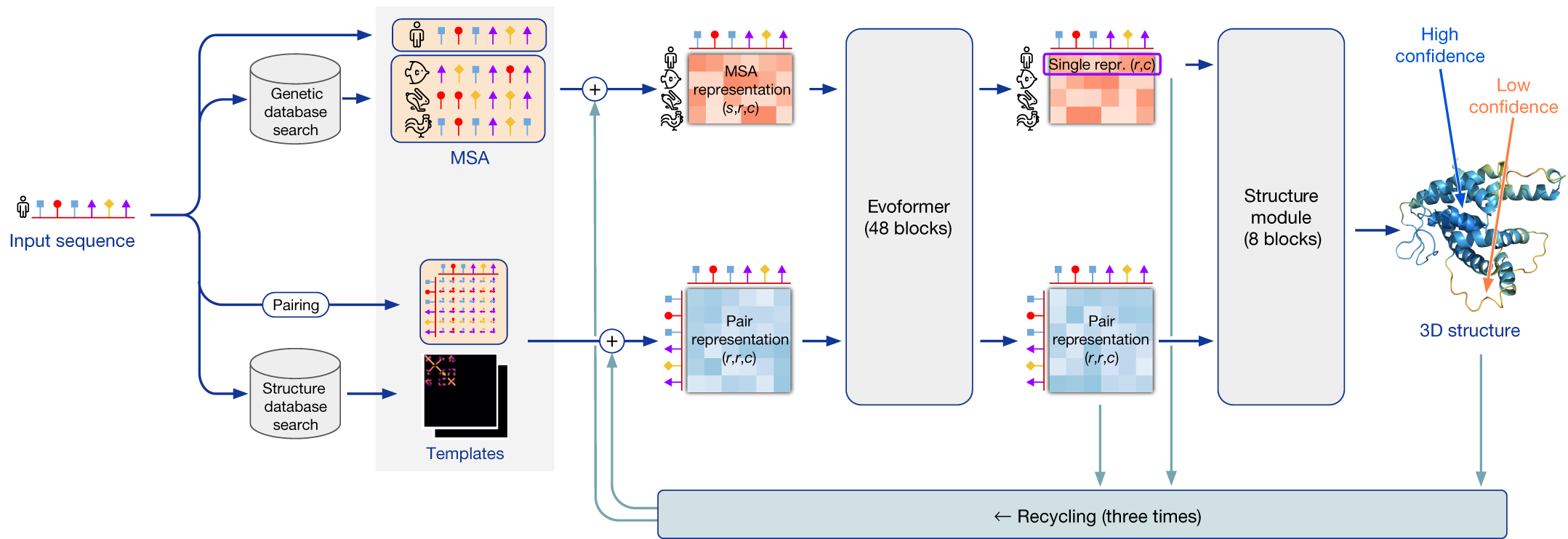
AlphaFold 方法示意图。 [@alphafold_approach_2021]
AlphaFold 蛋白质结构数据库¶
AlphaFold 预测模型的计算需要大量计算时间和资源(见AlphaFold 对生物化学领域的影响和AlphaFold 的工作原理)。 为了避免对相同蛋白质序列反复运行 AlphaFold,并促进 AlphaFold 蛋白质结构模型的传播和检查,DeepMind 团队与 EMBL 的欧洲生物信息学研究所(EMBL-EBI)合作创建了 AlphaFold 蛋白质结构数据库(AlphaFold DB)。 目前,该资源包含超过 2 亿个结构模型。 第一个 AlphaFold DB 发布版涵盖了人类蛋白质组,以及几个其他关键生物体,如拟南芥(Arabidopsis thaliana)和大肠杆菌(Escherichia coli)。 实际上,对于这些物种,其 UniProt 参考蛋白质组中的大多数蛋白质序列都由 AlphaFold 折叠。随后的发布扩展了包含的生物体列表。 最近的发布包含了几乎所有已知科学记录的蛋白质的预测结构,这将使 AlphaFold DB 扩大 200 倍以上——从近 100 万个结构到超过 2 亿个结构,覆盖了 UniProt 的大部分内容。 预计在未来几年,所有假设蛋白质都将添加到 AlphaFold DB。 这将包括目前被排除的病毒蛋白质。
AlphaFold DB 可以根据蛋白质名称、基因名称、UniProt 登录号或生物体名称进行搜索。 在实践作业中,你将学习如何使用 AlphaFold DB 以及如何将其纳入你的生物学发现流程中。 一个重要的剩余问题是:我们如何知道是否可以信任这些预测? 换句话说,我们如何知道是否可以相信 AlphaFold 预测的以及 AlphaFold DB 包含的三维结构模型?
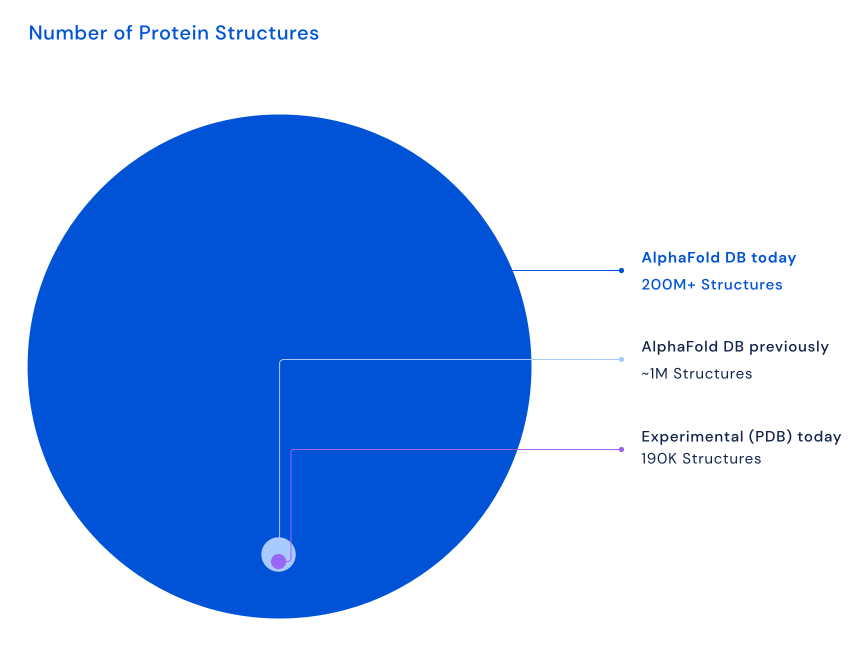
最近的发布包含了植物、细菌、动物和其他生物体的预测蛋白质结构,为研究人员利用 AlphaFold 推进其重要议题(包括可持续性、粮食不安全和被忽视疾病)的工作开辟了许多新机会。注意,PDB 包含实验验证的结构(目前约 218K),而 AlphaFold 产生的是预测结构模型。 [@alphafolddb_2022]
蛋白质结构模型质量¶
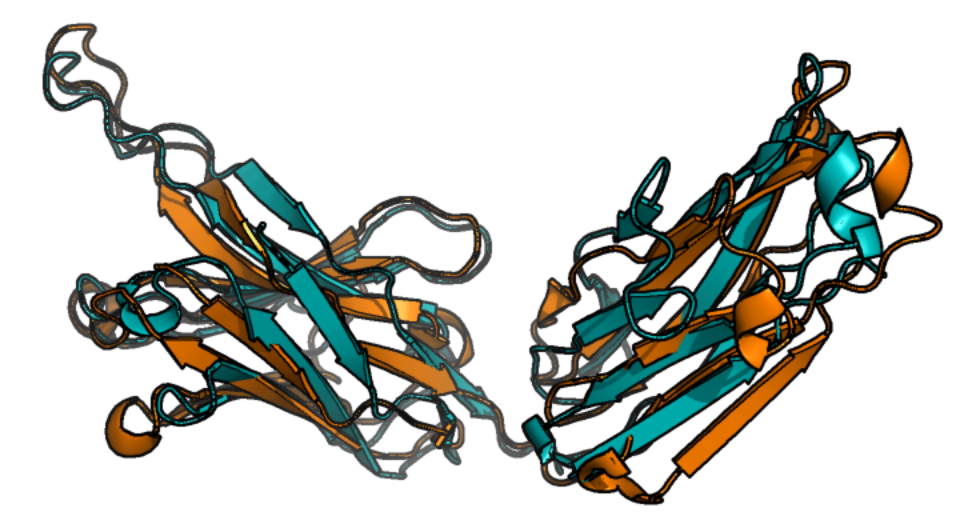
抗体晶体结构的重链部分(PDB:7MBF,橙色)与 AlphaFold 2 预测(蓝色)叠加。叠加视图展示了两个结构域的折叠在很大程度上被正确预测,其中三维蛋白质结构的某些部分比其他部分更好地拟合 PDB 结构。 [@blopig_2021]
预测模型只有产生某种置信度度量时才具有真正价值,因为如果没有关于预测确定性的任何概念,就很难解释结果并得出有意义的结论。 要了解预测与现实拟合的程度,需要将模型与真实情况进行比较。 下图展示了如何通过手动视觉检查两个叠加结构("真实"的实验获得的和预测的)来完成这一过程。 然而,要定量评估模型之间的差异,需要某种数值评分。在这里,我们将列出你本周将遇到的几个评分。 在本教材中,我们在 CASP 部分已经看到了三维蛋白质结构模型的一个比较度量:全局距离测试-总分(上图)。 你可能遇到的另一个评分是均方根误差(RMSE),基于α-碳的位置差异作为计算评分的输入。 原则上,模型的 RMSE 越小越好。 进行同源建模时,SWISS-MODEL 使用的 QMEAN-DISCO 评分用作质量度量。 该评分是多个指标的集合,共同提供模型质量的洞见。
要在没有与已知结构直接比较的情况下评估模型结构的置信度,需要评估氨基酸在三维坐标系中位置的不确定性。 AlphaFold 附带其自身的局部和全局误差预测,由机器学习模型计算(另见AlphaFold 的工作原理)。 局部误差关注氨基酸的个别位置,全局误差描述对不同蛋白质部分(可通过残基-残基相互作用)的预测置信度。 局部误差还用于在三维结构查看器中对模型的残基进行颜色编码。 这样,更容易观察结构模型中哪些部分比其他部分更可靠。 在实践作业中,你将更多地研究这两个不同的误差评分。
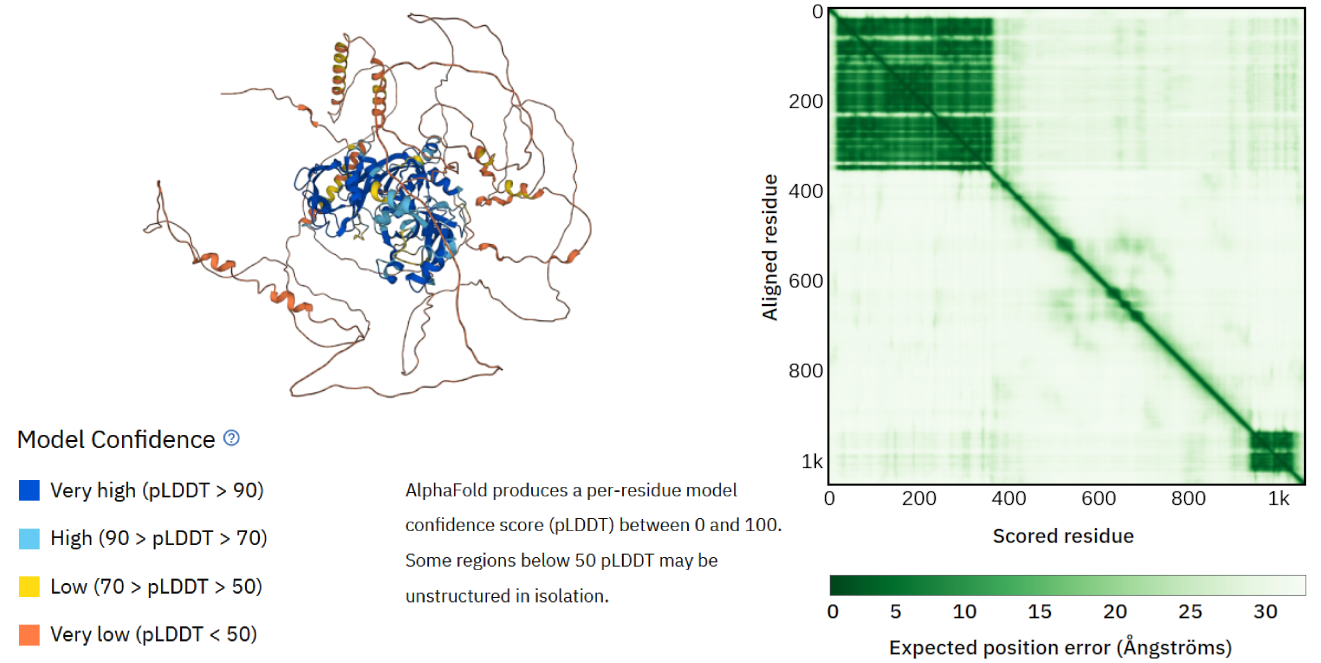
左图:生长素响应因子 16 的 AlphaFold 三维蛋白质结构模型。氨基酸残基根据局部置信度评分着色(另见 AlphaFold-PSD 和实践作业以获取进一步解释)。右图:AlphaFold 全局误差置信度评分概览。该视图显示蛋白质结构模型的两个部分误差较低(深绿色),其余结构误差较高——这些也对应于较低的局部置信度(另见 AlphaFold-PSD 和实践作业以获取进一步解释)。该结构模型可用于验证蛋白质的预测功能,并可作为进一步注释的起点,例如寻找其生物相互作用伙伴(即其他蛋白质、DNA、小分子)。 [@arf16_2022]
使用数据库时,你可能已经开始意识到它们也可能包含异常条目。 为了研究已知和预测的三维蛋白质结构的质量,可以使用拉氏图(Ramachandran plot)(第 1 章),例如检查是否有任何氨基酸残基位于拉氏图的非偏好区域。 在实践作业中,你将使用拉氏图。 这里需要注意的是,某些无序蛋白质只有在与其各种蛋白质伙伴存在时才进入有序排列;而其他蛋白质在任何条件下都不具有有序结构,这种特性可能对其功能至关重要。 如何最好地建模此类蛋白质的行为仍然是一个活跃的研究领域。
上述置信度度量也有助于突出预测方法的局限性。 在与 AlphaFold 相关的实践作业中,你将看到一些这方面的例子。 主要的教训是:你必须将预测视为预测:它是现实的模型,可能不能准确代表现实。 还要记住,蛋白质三维结构的某些部分我们自然可以更有信心。 例如,二级结构通常支撑三维蛋白质结构,而蛋白质中自然更无序的部分(如无规环)更难正确预测。 这些部分通常代表蛋白质结构在其生物环境中更灵活的部分,因此对(非常)灵活部分的任何预测都应被视为蛋白质结构的快照。 如果更详细地查看叠加的 PDB(实验性)三维结构和 AlphaFold 结构模型图像,你会看到这一点。在下图中,你可以看到预测模型对蛋白质中心较不灵活的部分更有信心,而对蛋白质外缘的灵活部分信心较低。
蛋白质结构模型:然后呢?¶
想象你生成了一个蛋白质结构模型,如上图中的那个。 你可以用它做什么?如上所述,它可以产生关于其可能的生化功能和作用的洞见。 换句话说,你可以开始形成可以在实验室中进行实验测试的假设。 你还可以开始预测蛋白质-蛋白质相互作用。 由于此类相互作用通常由三维结构元件(如裂缝、口袋等)驱动,这些元件通常作为活性位点,从序列预测此类三维结构元件将有助于更有信心地预测蛋白质-蛋白质相互作用。 此外,你已经看到在多序列比对中比较蛋白质序列如何有助于了解它们的进化和功能关系;通过使用三维结构模型作为输入,可以在结构层面进行类似的比较,即建立"功能同源性"。 我们越来越意识到结构比序列更保守;因此,(多重)结构比对在折叠或亚基层面可以提供对蛋白质关系的更深入视角。
Foldseek¶
最近的一个工具允许在合理时间范围内基于蛋白质结构输入进行基于结构的比对,即 Foldseek [@foldseek_2024]。 Foldseek 使用一种新颖的三维相互作用(3Di)字母表和极其快速的类 BLAST 序列搜索方法。通过这种方式,Foldseek 团队克服了在超过 2 亿个 AlphaFold 结构可用的情况下进行大规模基于结构的比较这一日益艰巨的任务。 例如,传统的基于结构的比对工具将约需要 1 个月来将一个结构与数据库中的 1 亿个结构进行比较。 在实践作业中,你将探索 AlphaFold 和 Foldseek 的组合如何用于探索感兴趣的蛋白质序列的可能功能。
三级结构预测展望¶
可以预期 AlphaFold 模型将继续发展。 例如,AlphaFold 3 中最近的增加是包括蛋白质、核酸、小分子、离子和修饰残基在内的复合物的联合结构预测(下图)。
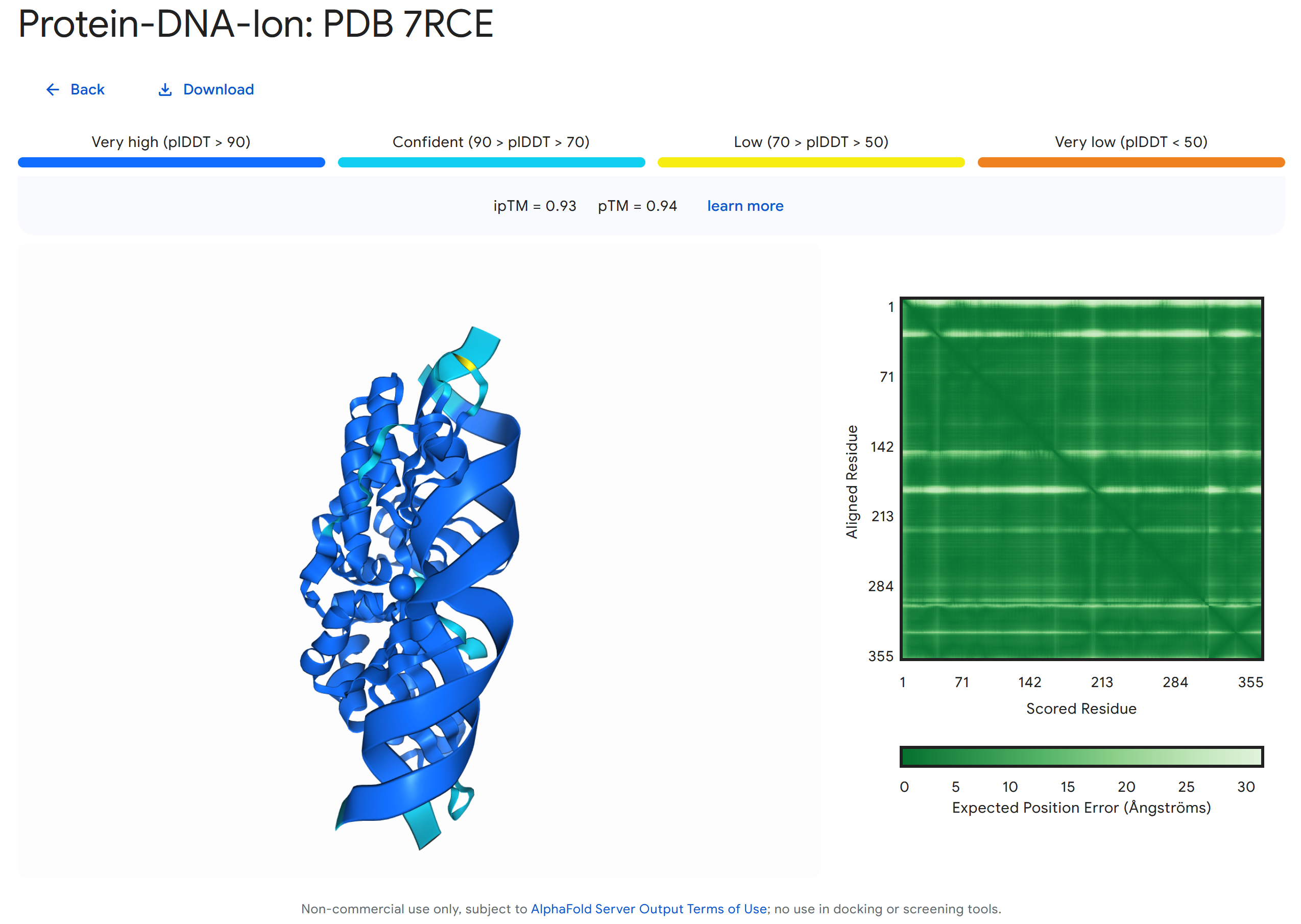
使用 AlphaFold Server 对 7RCE 蛋白与一段双螺旋 DNA 和两个离子(Ca²⁺ 和 Na⁺)相互作用的联合结构预测示例。 [@alphafold3_approach_2024]
这一最新趋势表明了结构预测从单一结构类型向多种结构类型的发展,以及从基于序列到基于结构的研究范式的转变。
另一个感兴趣的课题是蛋白质动力学建模。 许多蛋白质可以改变形状从而改变功能,例如取决于细胞条件,但这仍然很难建模。 最后,我们才刚刚开始探索翻译后修饰在为每个蛋白质生成许多(结构上、功能上)不同版本——即蛋白质组型(proteoforms)中的作用。 基于其当前表现,看看十年后该领域的发展方向将是令人兴奋的。 类似于我们在过去十年见证的手机-智能手机发展,到那时我们可能会对其能力感到惊讶。
Attention
AlphaFold 革命了三维蛋白质结构预测,并作为开发三维结构基工具(如 Foldseek)的催化剂,这些工具可以在大规模上比较蛋白质三维结构。AlphaFold 蛋白质结构数据库包含超过 2 亿个预测的三维蛋白质结构,该资源避免了在公共资源中的大多数蛋白质序列上运行 AlphaFold 的需要。AlphaFold 附带全局和局部误差预测,以提供三维结构模型的质量评估。
注释 4.1
最近,OpenFold Consortium 发布了 DeepMind 的 AlphaFold 2 的复现版本。这是一个开源的机器学习模型,在其 GitHub 上公开可用。 OpenFold 致力于向研究人员和商业公司提供最先进的基于 AI 的蛋白质建模工具,他们将能够使用、改进并为建模工具的开发做出贡献。 此类发展提高了快速发展的三级结构预测领域的 FAIR 性(第 1 章)。
实践作业¶
本实践包含问题和练习,帮助你处理第 4 章的学习材料。 你有 2 个上午的时间来完成这些练习。 在单个环节中,你应该争取完成大约一半的内容,即作业 I-III,最好进展到作业 IV 的一半。 这些实践练习为第 6 章的项目提供了最佳准备,工具及其使用方法也是考试内容的一部分。 因此,确保你现在培养实践技能,以便在项目中应用它们,并在考试中展示你的观察和解释能力。
注意,答案将在实践课后发布!
肽段折叠,20 分钟
你将简要回顾第 1 章,因为肽段折叠对蛋白质中任何二级和三级结构元件都是关键的。 完成本作业后,你将能够演示空间位阻如何在蛋白质折叠中起决定性作用。
访问 http://bioinformatics.org/molvis/phipsi/ 并等待应用程序加载。 使用侧面的单选按钮选择 φ 或 ψ 键,并增大或减小它们的角度,观察这如何影响三维表示。回答以下问题。
- 你如何在网络应用程序中识别肽键?
- 将 φ 和 ψ 接近 -175°。这种组合在拉氏图中是否处于"允许"区域?如果是,它可能折叠成什么结构?
- 为什么拉氏图的 x 和 y 轴刻度从 -180° 到 180°,而不是 0° 到 360°?
- 打开右侧的"显示空间冲突"(Show Clashes)。你能找出为什么拉氏图中 0°, 0° 区域如此空的原因吗?
- 尝试 φ 和 ψ 的不同组合。写下至少三种在拉氏图中不允许的 φ 和 ψ 组合,并使用这些角度折叠中心氨基酸。这种折叠是否真的不可能?注意,非键合原子通常应至少相距 ±2.5Å(其范德华半径之和),以避免空间位阻——在应用程序中以原子间彩色区域指示冲突发生的位置。
- 你能否推理上述观察如何得出空间位阻对蛋白质二级和三级结构元件重要的结论?
蛋白质结构搜索与可视化,50 分钟
蛋白质结构使用多种技术进行测量。 一旦发表,它们存储在**蛋白质数据库,或 PDB** 中。 核心数据库称为 wwPDB,有多个门户网站可以访问数据;其中 RCSB 提供的是最知名的。 在本作业中,我们将通过 RCSB 网站检查和可视化其中一些结构。 为了探索可用工具,我们将使用以下蛋白质的不同结构:人类泛素样蛋白 NEDD8、小鼠转录因子 STAT3 和海参珠蛋白。 完成本作业后,你将能够通过蛋白质数据库中的可视化来检查蛋白质三维结构。 1. 首先,在**UniProt** 中查找**人类**(!)NEDD8 蛋白,一种泛素样蛋白。泛素是一种小蛋白质(76 个氨基酸"AAs"),存在于许多细胞类型中(因此得名:无处不在)。它参与多种细胞过程的调控:蛋白质通过添加一个或多个泛素蛋白("泛素化")被标记为重定位、相互作用、降解等。它在真核生物中高度保守,其结构已被广泛研究。如果你找到了正确的条目,你会注意到提到了 81 个氨基酸。你能在页面上找到为什么蛋白质的总长度被标记为 81 个氨基酸吗?然后,通过左侧菜单转到"Structure"选项卡。该蛋白质有多少个螺旋和链? 2. 保持在"Structure"选项卡。在三维结构可视化下方,你看到蛋白质数据库(PDB)中许多可用的结构:一些仅包含一条泛素链,一些包含多条链。获得的最佳分辨率是多少?你认为该分辨率好吗? 3. 查找或筛选基于 NMR 的结构。在 RCSB PDB 中找到并打开 2N7K NMR 衍生的结构,并查看左上角的三维可视化(在 "Structure Summary" 选项卡上)。你在可视化中注意到了什么? 4. 返回 NEDD8 UniProt 页面,并在 RCSB PDB 中打开包含 4 条链的最高分辨率结构 1NDD。点击 "Structure" 选项卡。PDB 提供几个内联查看器——在三维视图和绘图选项的右下角选择 JSMol 查看器。你现在可以使用鼠标检查蛋白质结构(旋转、缩放)。在图像右侧,为"Assembly"选择"Asymmetric Unit",为"Color"选择"by Chain"。你观察到了什么?我们称这样的复合体为什么? 5. 按氨基酸残基的疏水性为复合体着色。查看器中似乎缺少颜色标尺,但你认为红色和蓝色在这里表示什么? 6. 将"Assembly"的可视化改回"Biological Assembly 1",使用"Cartoon"样式,并按"Secondary structure"着色。你能认出你在问题 1 中找到的结构元件吗? 7. 将查看器切换为 Mol*。通过将指针悬停在结构上,你可以获取每个残基及其在氨基酸序列中位置的信息。检查链中氨基酸的排列顺序,β-折叠片中的四条较长β-链是平行还是反平行的? 8. 将查看器切换为 JSmol,右键单击结构模型并找到"Style"选项——然后选择氢键并点击"calculate"。按照相同步骤查看任何二硫键(点击"on")。请参阅 Jsmol 文档获取帮助(查看器下方的帮助链接)。结果与你对二级结构的了解一致吗?你可以选择"Ball and Stick"样式来查看各个原子是如何连接的。 9. 在 RCSB PDB 中搜索另一个结构以使用 Mol* 查看器进行可视化,256B,并关注与血红素([HEM]109:B)的相互作用。有多少残基似乎参与了这种相互作用?涉及什么类型的相互作用?对于这样的结构/空间来说这现实吗? 10. 一些结构包含更大的配体,例如 DNA 序列。一个例子是 1BG1,一个转录因子 STAT3 的结构。使用 Mol* 查看器在 PDB 中查找它。请注意,如果你点击一个残基,它的相互作用也会显示出来,包括与(DNA)配体的相互作用。根据你对三维结构的检查,大约有多少核苷酸对似乎参与了蛋白质-DNA 结合?结合的首选核苷酸序列称为基序(motif);你可以在转录因子结合谱数据库 JASPAR 中查看 STAT3 的基序。 11. 从 1BG1 PDB "Structure Summary" 页面上的 "Literature" 框中,跟随主要引文"Search on PubMed"链接,找到首次描述 STAT3 结构的论文。在该论文的图 3 中,DNA 结合位点看起来像你在问题 10 中找到的那个吗? 12. 作为最终演示,在 RCSB PDB 中转到海参珠蛋白的结构 1HLM,并在"Structure Summary"下的"Macromolecules"部分中,通过三维结构找到相似蛋白质(点击链接)。它与哪个其他结构(部分)最相似?通过点击两个蛋白质条目并在窗口右上角的"Download Files"下拉菜单中选择"PDB format"来下载 1HLM 和最相似结构的 PDB 文件。然后,使用 TM-score 对它们进行比较。你从输出中能得出什么结论,这些结构相似吗?哪些指标表明了这一点?
二级结构预测,50 分钟
1TIM 是一个典型的 TIM 桶 PDB 结构,属于磷酸丙糖异构酶蛋白。 TIM 桶存在于许多蛋白质中,并在蛋白质折叠数据库 CATH 中形成一种拓扑结构(折叠)(在作业 V 中你将获得折叠数据库的实践经验)。 查看 Wikipedia 了解更多关于 TIM 桶的信息,并弄清楚为什么该结构被称为"桶"。 完成本作业后,你将能够解释将蛋白质二级结构预测器应用于蛋白质序列的结果。 1. 首先,让我们研究 1TIM 的二级结构指派工具 DSSP、PALSSE 和 Stride 的结果。这些工具基于三维蛋白质结构预测二级结构元件。你可以在 BrightSpace 上第 4 章 Practicals_Files 下的 pdf 文件中找到这些工具的结果。检查三种方法预测为螺旋、折叠和其他的残基比例。DSSP 和 STRIDE 的结果与 PALSSE 的结果之间的主要区别是什么?什么可以解释这种差异? 2. 检查结果文件中的结构比对。这是否确认了你在问题 1 中假设的差异原因? 3. 根据你的结果,你认为有多少个螺旋?多少个折叠片?这与 Wikipedia 中的信息匹配吗? 4. 为了评估二级结构预测的质量,有几种评分指标。最直接和最容易计算的是 Q3 评分。Q3 值是正确预测的残基在二级结构状态(即螺旋、链和卷曲)中的比例。你可以在图中找到更多信息。手动计算 DSSP 对链 A 预测的 Q3,假设共识为实际二级结构(即"真实标签")。 5. 为了获取更多关于蛋白质属性的信息,你可以通过在 服务器 上传相应的蛋白质序列(UniProt ID P00940)(选择完整的 2-248),预测所研究的 1TIM 三维结构的局部疏水性(Kyte-Doolittle,在 9 个残基窗口上平均)。将输出与问题 a 的 DSSP 二级结构预测进行比较。你预期核心是由螺旋还是折叠片组成?你是如何得出答案的?如何验证这一点? 6. 我们可以使用最先进算法 NetSurfP-3.0 预测二级结构。将磷酸丙糖异构酶蛋白(P00940)的 FASTA 蛋白质序列复制粘贴到指定字段(或上传 FASTA 文件),NetSurfP-3.0 使用一种将蛋白质序列视为语言句子的机器学习方法。通过基于 PDB 中已知蛋白质三维结构及其 DSSP 计算属性训练大型模型,NetSurfP-3.0 从一级序列预测二级结构属性。将多个序列的信息组合用于二级结构预测有什么优势? 7. 根据 NetSurfP-3.0 结果页面,图形概览中 SS3 结果显示的螺旋和折叠片数量与你在问题 3 中的先前答案相比如何?图片与问题 1 的结果相比如何? 8. 通过将指针悬停在图形概览中的残基上,研究第 45个残基(丝氨酸)的 SS3 和 SS8 预测。你能推测为什么 SS3 预测为"卷曲"而 SS8 预测为"310 螺旋"吗?为了进一步探索这一现象,我们将查看 2StructCompare 的结果文件,通过 BrightSpace(Practicals_Files)获取,并结合 PDB 中显示的三维结构使用 Mol* 查看器。如果你查看这些截图和三维查看器,你认为该残基是螺旋还是卷曲?此外,调查可用截图和三维查看器中约第 100 个氨基酸残基处第 4个螺旋的"分裂"。你认为这是分裂吗? 9. NetSurfP-3.0 同时预测相对表面可及性(结果页面图形视图中的顶部线条)。结果是否与你在 5. 和 8. 中的发现一致? 10. 最后,我们将进行跨膜区预测。让我们首先使用 TMHMM 预测 SigmaR1(UniProt ID Q99720)、水通道蛋白(P47865)和 1TIM(P00940)的跨膜拓扑结构。对于每个蛋白质,从 UniProt 复制/粘贴 FASTA 序列到 TMHMM 并按"Submit"。考虑到蛋白质描述和可用的三维结构,预测是否合理? 11. 使用较新版本的 TMHMM DeepTMHMM 对水通道蛋白(P47865)和 1TIM(P00940)执行与之前相同的操作。结果与 10. 中找到的相比如何?
AlphaFold 三维结构预测,50 分钟
大多数蛋白质结构建模方法在计算上非常密集,在本实践中尝试将花费太多时间。 目前这仅适用于自动化同源建模(作业 VI),但首先我们将通过 AlphaFold 数据库和通过 CATH 数据库的功能域来研究 AlphaFold2。 完成作业 IV 后,你将能够在 AlphaFold 数据库中搜索预测的三维蛋白质结构模型,并在局部和全局层面评估这些模型的质量。
AlphaFold 的引入彻底改变了蛋白质结构预测领域。 在这里,我们将通过 AlphaFold 蛋白质结构数据库 评估预计算的现成 AlphaFold2 模型,该数据库目前提供 >200,000,000 个结构模型。 1. 咖啡植物(Coffea arabica)对许多人类来说是重要的作物植物。访问 AlphaFold 蛋白质结构数据库 并搜索生物体 Coffea arabica。你能找到多少个匹配?这与植物模式生物拟南芥(Arabidopsis thaliana)和人类基因组相比如何? 2. 咖啡以其咖啡因闻名。在新标签页中打开来自 Coffea arabica 的"Probable caffeine synthase MTL3"AlphaFold 结构预测页面(AlphaFold DB ID:A0A096VHX6)并研究该结构模型。放大结构模型的各个部分,点击残基以显示氨基酸骨架和侧链及其相互作用。特别是研究该结构模型的局部"逐残基置信度评分"(pLDDT)。该模型的准确性如何?哪些部分最准确?这与你的先验知识一致吗? 3. "Probable caffeine synthase MTL3"的生物学功能是推测性的。跟随到 UniProt 页面的链接。它在咖啡因生物合成中的可能作用是什么? 4. AlphaFold 的出现和由此产生的 AlphaFold 数据库推动了其他工具的发展。正如你在教材中所读到的,蛋白质功能主要由其结构而非序列决定。随着三维结构的可获得性增加,特别是预测的结构,现在可以使用预测或实验三维结构作为输入,基于折叠搜索相关蛋白质。返回"Probable caffeine synthase MTL3"的 UniProt 页面,转到"Structure"选项卡。在 AlphaFold 模型视图下,实际 AlphaFold 模型旁边有指向 Foldseek 的链接。点击该链接,在 Foldseek 中打开一个预填充了预测"Probable caffeine synthase MTL3"模型结构的新搜索并开始搜索。基于"all databases"选项卡中的结果,你可以推断出什么功能?点击右侧的对齐按钮查看一些匹配。它们看起来如何?由于列表顶部主要由预测结构占据,转到"PDB100"选项卡(滚动到屏幕顶部的右侧——这仅包含来自 PDB 的条目),并执行相同操作。你在什么类型的生物体中找到了匹配?匹配的序列相似性如何,与结构相似性的关系如何? 5. 现在新标签页中打开 Coffea arabica 的"DNA-directed RNA polymerase subunit alpha"(UniProt A0A367)AlphaFold 页面。通过点击箭头研究全局"Predicted aligned error tutorial"。一旦你理解了误差模型,研究该结构模型的局部和全局误差模型。放大结构模型的感兴趣部分,点击一些残基——你看到了什么?关于结构模型质量你得出什么结论? 6. 现在,考虑以下情况:一位同学有一个来自植物样本的蛋白质序列,她想要表征它:
MREGRETKNGNGHVGRRASSQVWEFDPGDPDELVVVAEAARRGFVVRRHELKHSSDLL
MRMQFAKANPLKLDIPAIKLEEHEAVTGEAVLSSLKRAIARYSTFQAHDWPGDYGGPM
FLMPGLIITLYVSGALNTALSSEHQKEIRYLYNHEDGGWGLHIEGHSTMFGGSALTYV
SLRLLGEGPDSGDGAMEKGRKWILDHGGATYITSWGKFWLSVLGVFDWSGNNPVPPEI
WLLLLPYFLPIHPGRMWCHCRMVYLPMCYIYGKRFVGPVTPIILELRKELYEVPYNEV
DWDKARNLCAKEDLYYPHPFVQDVLPATLHKFVEPAMLRWPGNKLREKALDTVMQHIH
YEDENTRYICIGPVNKVLNMLACWISEAFKLHIPRVHDYLWIAEDGMKMQGYNGAFTV
QAIVATGLIEEFGPTLKLAHGYIKKTQVIDDCPGDLSQWYRHISKGAWPFSTADHGWP
ISDCTAEGLKAALLLSKISPDIVGEAVEVNRLYDSVNCLMSYMNDNGGFAIRPTELLL
TRSYAWLELINPAETFGDIVIDYPYVECTSAAIQALTAFKKLYPGHRKSEIDNCISKA
ASFIEGIQKSDGSWYGSWAVCFTYGTWFGVKGLVAAGRTFKNSPAIRKACDFLLSKEL
PSGGWGGESYLSSQDQVYTNLEGKRPHAVNTGWAMLALIDAGQAERDPIPLHRAAKVL
INLQQSEDGEFPQQEIIGVFNKNCMISYSEYRNIFPIWALGFAIRDATAWISE
 对 Uniprot + alphafold 数据库运行 blastp 的最佳 BLAST 匹配。 [@alphafold3_approach_2024] 呈现的匹配对你来说有意义吗?是否有你认为足够好的匹配可以从中推断其模型的结构?研究 AlphaFold 模型。质量如何?该蛋白质的可能功能是什么? 7. 返回"Probable caffeine synthase MTL3"AlphaFold 结构预测页面。根据你对全局预测对齐误差模型的新知识,你现在对该模型有什么看法? 8. 有人说 AlphaFold 将解决我们在蛋白质结构折叠方面的所有问题......但现在下这个结论可能还为时过早。查看人胰岛素(UniProt P01308)的结构模型?提出的模型质量如何?你能想到原因吗?
对 Uniprot + alphafold 数据库运行 blastp 的最佳 BLAST 匹配。 [@alphafold3_approach_2024] 呈现的匹配对你来说有意义吗?是否有你认为足够好的匹配可以从中推断其模型的结构?研究 AlphaFold 模型。质量如何?该蛋白质的可能功能是什么? 7. 返回"Probable caffeine synthase MTL3"AlphaFold 结构预测页面。根据你对全局预测对齐误差模型的新知识,你现在对该模型有什么看法? 8. 有人说 AlphaFold 将解决我们在蛋白质结构折叠方面的所有问题......但现在下这个结论可能还为时过早。查看人胰岛素(UniProt P01308)的结构模型?提出的模型质量如何?你能想到原因吗? 自动化同源建模,45 分钟
现在,我们将转向自动化同源建模。 完成本作业后,你将能够创建自动化同源三维蛋白质结构模型,并通过检查模型和模板三维结构来评估模型质量。 由于 Q5YGP8 蛋白没有近缘结构同源物,我们将在本作业中使用另一种蛋白质:UniProt ID Q8QGC7。 1. 首先,在 UniProt 中查找该蛋白质。它是什么类型的蛋白质?有多少个残基? 2. 使用 BLAST 在 UniProt 中搜索具有(实验性!)三维结构的相似蛋白质(确保选择了正确的目标数据库!!)。你找到了与下方呈现的结果相似的匹配吗?在什么长度和一致性下?你认为这些结构是否足够好以用作同源建模中的模板?注意,如果 BLAST 运行未能及时完成,你可以使用下方粘贴的结果。  3. 由于似乎有合适的模板,我们可以尝试自动化同源建模。访问 SWISS-MODEL,粘贴 FASTA 序列,选择"Search for Templates",然后去泡杯咖啡。等你回来时,找到了多少个模板?它们是唯一的吗? 4. 选择"Sequence similarity"选项卡,获取我们的搜索序列与数据库中序列之间关系的图形表示;通过点击每个相似序列,你可以看到比对在结构中的哪个部分。你注意到了什么? 5. 在左侧"Sequence similarity"选项卡下的图形视图中,选择几个模板,并选择"Alignment"选项卡以查看比对。比对质量好吗? 6. 返回"Templates"选项卡,选择几个匹配良好的 PDB(!)来源模板(即 6KPG、6N4B、5TGZ、7WV9 等)和至少一个匹配较差的模板(如 5DYS、2R4S 等)。点击"Build Models"并等待结果;同时,阅读关于如何解释结果的文档。在质量图中,什么颜色表示"好",什么颜色表示"差"? 7. 你认为模型质量如何?检查 GMQE、QMEANDisCo 和 QMEAN Z-Score 值,以及质量图。 8. 打开模型-模板比对(通过点击每个模型结果框中的"v"形按钮)。通过将光标移到序列比对上,你可以可视化残基在三维结构中的位置(反之亦然)。哪里拟合最差?这是否与结果顶部"Local quality"图一致?你能解释这一点吗?提示:你还可以通过点击右侧三维图下方的比对来叠加模型可视化。 9. 最后,通过按左侧每个模型下的"Structure assessment"按钮并研究拉氏图,在 SWISS-MODEL 中评估"好"和"差"三维模型的结构质量。将拉氏图与模板的拉氏图进行比较,你可以在 PDBSum 中查看。你发现了什么? 10. 一旦蛋白质结构已知,它们就可以用来了解其相互作用和功能。作为示例,在 PDB 和 PDBsum 中检查蛋白质 3i49。该蛋白质催化什么反应?需要什么辅因子?在 PDB 中可视化配体相互作用,并在 PDBsum 中使用 Ligand/Metal 选项卡了解更多关于该结构的信息。哪些残基构成活性位点?这告诉你关于序列-结构-功能范式的什么? 11. 现在,返回 Q8QGC7 的 UniProt 页面。你可能记得在 AlphaFold 作业中,我们期望在这里也有一个 AlphaFold 预测的三维结构模型。确实,在"Structure"选项卡下,有一个预测结构!它与 6. 中同源建模预测的结构相比如何?
3. 由于似乎有合适的模板,我们可以尝试自动化同源建模。访问 SWISS-MODEL,粘贴 FASTA 序列,选择"Search for Templates",然后去泡杯咖啡。等你回来时,找到了多少个模板?它们是唯一的吗? 4. 选择"Sequence similarity"选项卡,获取我们的搜索序列与数据库中序列之间关系的图形表示;通过点击每个相似序列,你可以看到比对在结构中的哪个部分。你注意到了什么? 5. 在左侧"Sequence similarity"选项卡下的图形视图中,选择几个模板,并选择"Alignment"选项卡以查看比对。比对质量好吗? 6. 返回"Templates"选项卡,选择几个匹配良好的 PDB(!)来源模板(即 6KPG、6N4B、5TGZ、7WV9 等)和至少一个匹配较差的模板(如 5DYS、2R4S 等)。点击"Build Models"并等待结果;同时,阅读关于如何解释结果的文档。在质量图中,什么颜色表示"好",什么颜色表示"差"? 7. 你认为模型质量如何?检查 GMQE、QMEANDisCo 和 QMEAN Z-Score 值,以及质量图。 8. 打开模型-模板比对(通过点击每个模型结果框中的"v"形按钮)。通过将光标移到序列比对上,你可以可视化残基在三维结构中的位置(反之亦然)。哪里拟合最差?这是否与结果顶部"Local quality"图一致?你能解释这一点吗?提示:你还可以通过点击右侧三维图下方的比对来叠加模型可视化。 9. 最后,通过按左侧每个模型下的"Structure assessment"按钮并研究拉氏图,在 SWISS-MODEL 中评估"好"和"差"三维模型的结构质量。将拉氏图与模板的拉氏图进行比较,你可以在 PDBSum 中查看。你发现了什么? 10. 一旦蛋白质结构已知,它们就可以用来了解其相互作用和功能。作为示例,在 PDB 和 PDBsum 中检查蛋白质 3i49。该蛋白质催化什么反应?需要什么辅因子?在 PDB 中可视化配体相互作用,并在 PDBsum 中使用 Ligand/Metal 选项卡了解更多关于该结构的信息。哪些残基构成活性位点?这告诉你关于序列-结构-功能范式的什么? 11. 现在,返回 Q8QGC7 的 UniProt 页面。你可能记得在 AlphaFold 作业中,我们期望在这里也有一个 AlphaFold 预测的三维结构模型。确实,在"Structure"选项卡下,有一个预测结构!它与 6. 中同源建模预测的结构相比如何?
项目准备练习
本作业是一个类似项目的问题:提醒你,区别在于你不会被引导到需要使用的确切工具,而是我们期望你使用教材和实践作业中的知识和专业知识来找到合适的工具。 换句话说,本作业旨在让你探索在作业 I-V 中访问过的工具(或你可能找到的替代工具),并对所研究蛋白质的结构和功能得出相关观察结果。 完成本作业后,你将能够基于蛋白质的序列和结构,调查已知属性并预测可能属性。
我们继续探索拟南芥(Arabidopsis thaliana)中 ARF 基因家族的成员。 你会记得 ARF5(UniProt ID P93024)和 IAA5(UniProt ID P33078)是两个研究较多的 A. thaliana 蛋白,它们在生长素介导的基因表达调控中发挥作用。
正如你现在可能意识到的,本章全部关于结构和功能! - 简要探索 ARF5 和 IAA5 的三维结构——结构如何支持功能?哪些二级结构元件重要?什么驱动了 ARF 和 IAA 之间的相互作用?你可以找到哪些结构域?你能了解它们的什么功能? - 对 ARF5 和 IAA5 执行二维结构预测,并将结果与三维结构进行比较。 - 在 UniProt 中选择一个在 PDB 中没有实验性三维结构的 ARF 或 IAA。按照描述的生物学发现流程,使用二级和三级结构预测来描述该序列。你能报告什么?
为上述三点撰写简短的要点式笔记(请注意,在项目中,你需要撰写简短的文本块而非要点)。 你可能想用你访问的工具的截图来突出一些发现——本作业最多使用 6 张图。 1. 材料与方法 你做了什么?你使用了哪些数据、数据库和工具,为什么选择这些?你选择了哪些重要设置? 2. 结果 你发现了什么,主要结果是什么?报告相关数据、数字、表格/图片,并清楚描述你的观察结果。 3. 讨论与结论 思考你发现的结果是否有意义。它们是否符合你的期望,还是你看到了令人惊讶的东西?结果意味着什么,你如何解释它们?不同工具的结果是否一致?你能得出什么结论?确保描述你的解释所基于的预期和假设。
我们鼓励你与同学、助教和老师讨论你的结果。 通常,他们可能发现了不同但互补的信息,在一起你们能够更完整地描绘蛋白质家族的全貌。
参考文献¶
{{ bibliography }}
术语表¶
从头计算方法(Ab initio approach) 计算量大的三维蛋白质结构预测方法,需要人类专家输入,使用氨基酸残基及其环境的理化性质来模拟蛋白质序列的折叠过程。
AlphaFold 基于机器学习的三维蛋白质结构预测方法,使用在整个蛋白质数据库上训练的深度学习模型。
AlphaFold 蛋白质结构数据库(AlphaFold DB) 包含超过 2 亿个 AlphaFold 预测的三维蛋白质结构模型的资源。
蛋白质结构预测关键评估(CASP) 全社区范围的竞赛,要求研究小组预测没有任何公开三维结构的蛋白质序列的三维结构。最近扩展了额外挑战,如蛋白质-蛋白质相互作用和蛋白质-配体相互作用的预测。
DeepTMHMM/TMHMM 使用深度学习模型(DeepTMHMM)或隐马尔可夫模型(TMHMM)预测蛋白质中跨膜区的工具,将蛋白质序列作为输入,然后输出相应的逐残基标签。
预期位置误差(Expected position error) AlphaFold 预测的全局误差度量,用于指示氨基酸残基位置相对于所有其他残基的置信度。
Foldseek 基于三维相互作用(3Di)字母表进行大规模结构-结构蛋白质比对的工具。
全局距离测试-总分(GDT-TS) CASP 使用的三维蛋白质结构预测评估指标:预测结构中氨基酸的α-碳在最佳可能比对下,在埃阈值距离内的百分比。
同源建模(Homology modelling) 使用实验测量的蛋白质结构的模板结构的三维蛋白质结构预测方法。
NetSurfP 使用深度学习模型预测α-螺旋和β-链等二级结构元件的机器学习方法。
标注(Labelling) 为数据点添加已知类别,例如氨基酸的结构状态,即α-螺旋、β-链或"无规卷曲"。
蛋白质序列-结构-功能范式(Protein sequence-structure-function paradigm) 该理论假设氨基酸序列原则上包含描述其三维结构的所有信息,且蛋白质三维结构原则上包含定义其功能的所有信息。
均方根误差(RMSE) 评估两个三维蛋白质结构比较质量的评分,使用氨基酸残基α-碳的位置差异。
SignalP 基于机器学习的工具,用于预测和区分不同类型的信号肽。
信号肽(Signalling peptide) 细胞转运机制的肽识别信号,用于将蛋白质运输到其活性位置,即细胞膜或内质网。
逐残基模型置信度评分(plDDT) AlphaFold 预测的局部误差度量,用于指示氨基酸残基位置相对于一级序列中相邻残基的置信度。
蛋白质数据库(PDB) 包含实验测量的蛋白质三维结构的资源。
穿线法(Threading approach) 预测蛋白质短片段或折叠的三维结构的预测方法,又称基于片段的方法。
跨膜区(Transmembrane section) 蛋白质序列中跨越细胞膜的部分。
跨膜蛋白(Transmembrane protein) 至少跨越细胞膜一次的蛋白质。
三个区域(蛋白质三级结构预测的) 基于查询蛋白质序列与数据库条目之间的序列一致性百分比与比对长度的图,可以定义三个"区域"。根据查询序列的最佳匹配落入哪个区域,需要不同的传统方法来预测(部分)其三维结构。
本文阅读量 次本站总访问量 次