第一章
课程来源与翻译声明
- 原著版权:本教程翻译自瓦赫宁根大学(Wageningen University & Research)的经典开源课程 Introduction to Bioinformatics。
- 翻译说明:本版本主要采用人工智能(机器翻译)进行全文本地化,并由团队进行了初步校对。部分专业术语可能存在翻译不够地道或准确的地方。
- 勘误反馈:若你在学习过程中遇到语病、错别字、代码失效或概念歧义,欢迎随时点击页面右上角的 :material-edit: 编辑此页(或联系课题组/在 GitHub Issue 中)提交反馈,帮助我们持续完善本教程。
在本章中,你将学习理解生物信息学方法所需的分子生物学基础知识。此外,你还将学习存储和描述生物分子数据的常用方法。
学习目标
学习本章后,你应该能够: - 描述 DNA 和 RNA 序列的化学组成和结构差异,以及这些序列如何存储、复制和传递遗传信息。 - 描述遗传信息如何被转录和翻译为蛋白质——即中心法则。 - 解释氨基酸的理化性质如何驱动蛋白质的折叠和功能,以及氨基酸替换的影响。 - 描述基因组注释和基因预测的概念,以及它们如何结合计算方法和生物学证据来解码基因组内容。 - 列出组织和存储生物序列及注释数据的重要数据库和文件格式。 - 描述标准化本体 (Ontology) 如何帮助构建生物学元数据,并确保数据的互操作性和可重用性。
生物学背景¶
生物信息学的很大一部分涉及对生物学序列 (Sequence) 的分析。这些序列来源于在细胞中发挥重要作用的有机大分子。在本章的第一节中,我们将描述这些大分子、它们的序列,以及生成其活性结构和维持这些结构所涉及的生物学过程。
因此,本节为整个课程提供了重要的背景材料。根据你的背景,本节的部分内容可能看起来有些多余,在这种情况下,本节可以作为复习材料。后续章节假定你已经熟悉本节内容。
核酸¶
脱氧核糖核酸 (DNA) 携带着生物体的遗传信息。核糖核酸 (RNA) 参与蛋白质 (Protein) 的表达,也是某些病毒的遗传物质。因此,这些分子作为地球上生命的基础至关重要。基因组 (Genome) 指的是细胞 (Cell) 的全部遗传内容,而基因组学 (genomics) 是对基因组的研究。
DNA 和 RNA 由称为核苷酸 (Nucleotide) 的单体组成,每个核苷酸由三个组分构成:
- 戊糖,其中碳原子编号为 1' 到 5'(将 1' 读作"一撇")。戊糖的类型区分了 RNA 和 DNA:DNA 中的糖是脱氧核糖,而 RNA 中的糖是核糖。它们在结构上相似,但脱氧核糖在 2' 位置上是一个 H 而不是 OH。
- 磷酸基团,连接在糖的 5' 位置上。
- 碱基,连接在糖的 1' 位置上。
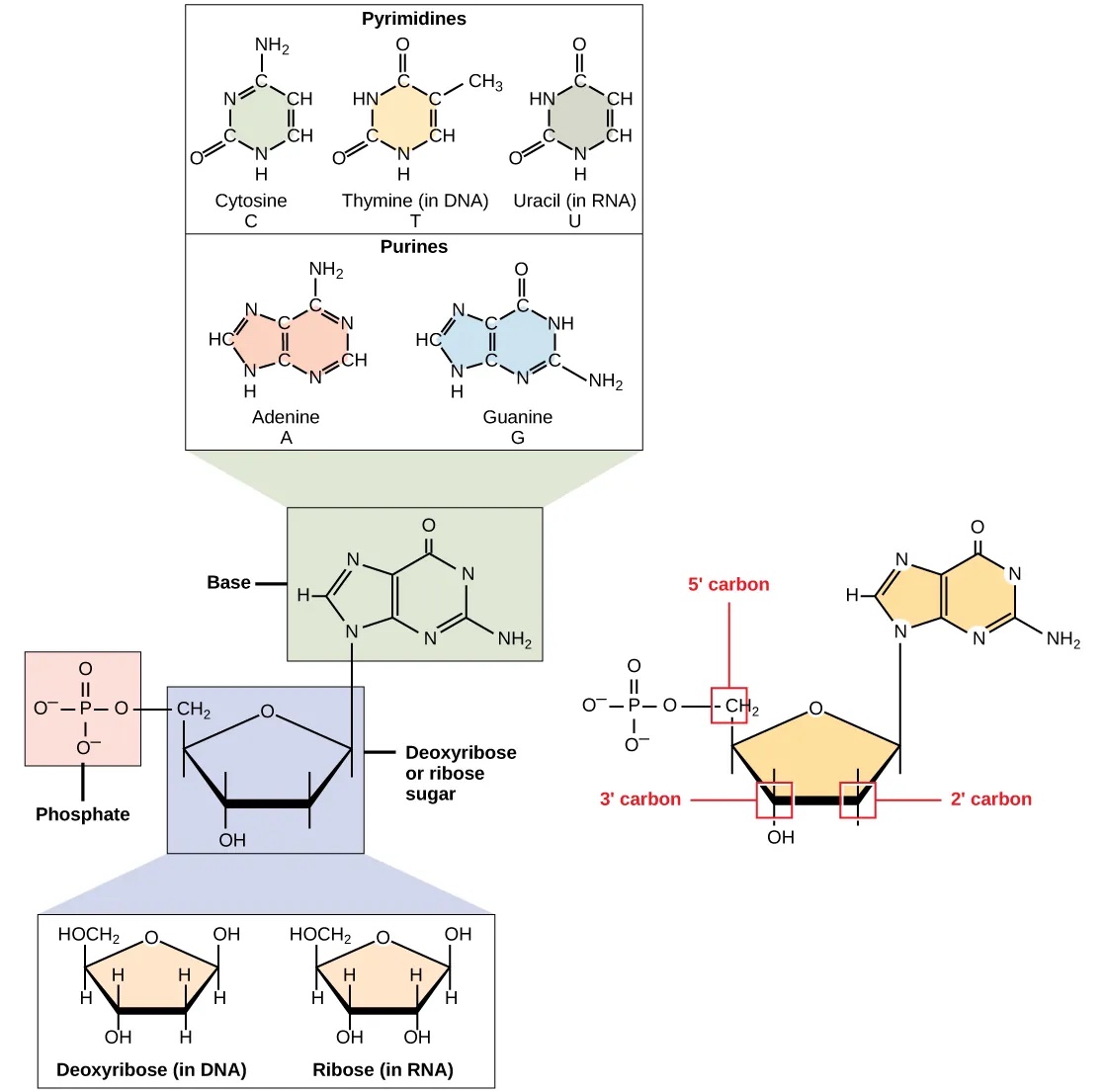 图:核苷酸的组成部分。 许可:CC BY 4.0 [@nucleotide_2018]
图:核苷酸的组成部分。 许可:CC BY 4.0 [@nucleotide_2018]
碱基可以分为两类:嘌呤(双环结构)和嘧啶(单环结构)。DNA 包含 A、T、C 和 G;而 RNA 包含 A、U、C 和 G。
核苷酸
核苷酸是所有生命中的核心分子。你不需要记住确切的化学结构,但需要了解 DNA 和 RNA 之间的区别、不同的碱基及其类别(嘌呤或嘧啶)。
DNA 双螺旋¶
DNA 分子是脱氧核糖核苷酸的聚合物,形成右手双螺旋。糖和磷酸位于外侧形成螺旋的骨架,碱基堆叠在内部并通过氢键相互结合。因此 A 通过两个氢键与 T 配对,C 通过三个氢键与 G 配对,它们是**互补**碱基。这些配对也称为 Watson-Crick 碱基配对,以 DNA 的发现者命名。
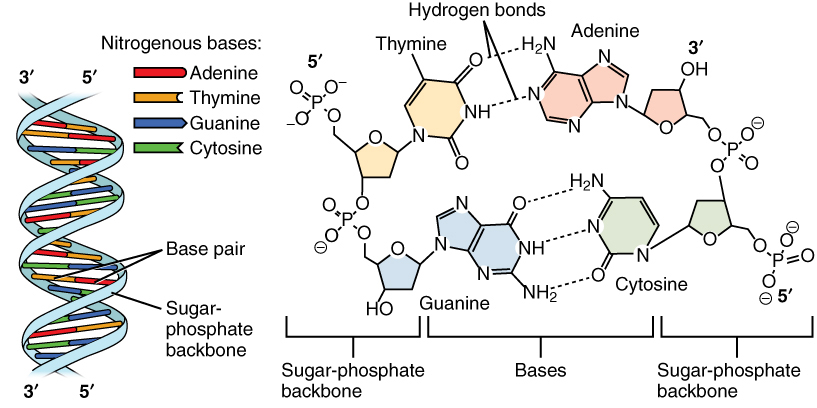 图:DNA 结构。 许可:CC BY 3.0 [@dna_alt_2013]
图:DNA 结构。 许可:CC BY 3.0 [@dna_alt_2013]
双螺旋的两条链以相反方向排列,也称为反平行,即一条链从 5' 到 3',另一条从 3' 到 5'。核苷酸序列通常以 5' 到 3' 方向书写。由于互补性,一条链的碱基序列可以从另一条链的碱基序列推导出来。这称为**反向互补**。例如,AAGT 的反向互补是 ACTT,其中两条链均以 5' 到 3' 方向给出。
DNA 复制¶
由于两条 DNA 链仅通过氢键连接,它们可以相对容易地被分离,例如在 DNA 复制过程中。分离的链各自作为模板,由 DNA 聚合酶以 5' 到 3' 方向合成新的互补链。这种复制模式称为半保留复制。
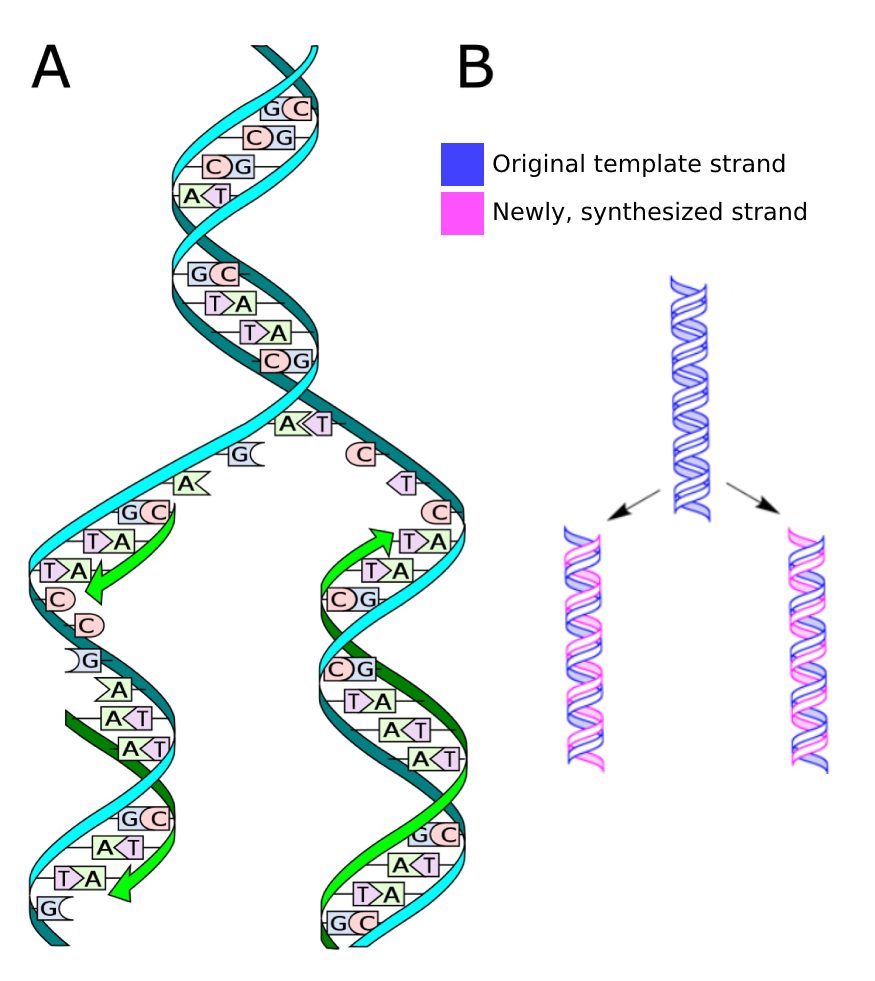 图:A)DNA 复制过程。许可:CC0 1.0 [@replication_a_2013]。B)半保留 DNA 复制,两个拷贝各包含一条原始链和一条新链。许可:CC0 1.0 修改自 [@replication_b_alt_2009]。
图:A)DNA 复制过程。许可:CC0 1.0 [@replication_a_2013]。B)半保留 DNA 复制,两个拷贝各包含一条原始链和一条新链。许可:CC0 1.0 修改自 [@replication_b_alt_2009]。
DNA 复制的错误率非常低,大约每 109 个碱基中只有一个错误碱基。这一特性在细胞分裂和世代传递过程中保护了遗传信息。同时,它也在进化时间尺度上导致突变的发生,我们将在后文(替换)中看到。
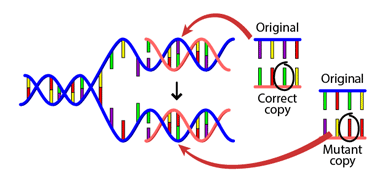 图:复制过程中发生的 DNA 突变。 许可:BY-NC-SA 4.0 [@dna_mutation_2020]
图:复制过程中发生的 DNA 突变。 许可:BY-NC-SA 4.0 [@dna_mutation_2020]
RNA、转录与剪接¶
在转录 (Transcription) 过程中,RNA 聚合酶以 3' 到 5' 方向读取模板链(也称为非编码链)。这产生了一条从 5' 到 3' 的 RNA 分子,它是编码链的拷贝。在转录过程中,胸腺嘧啶被尿嘧啶取代。与 DNA 不同,RNA 不形成稳定的双螺旋。RNA 主要是单链的,但大多数 RNA 显示出互补碱基之间的分子内碱基配对。
RNA 有四种主要类型:
- 信使 RNA (mRNA):稍后将翻译为蛋白质的 RNA 分子,因此在蛋白质生产中充当"信使"。
- 核糖体 RNA (rRNA):核糖体的主要组成部分(细胞的"动力工厂")。
- 转运 RNA (tRNA):作为"适配分子"发挥作用,在翻译过程中充当 mRNA 和蛋白质氨基酸序列之间的物理连接。
- 微小 RNA (miRNA):长度为 21-23 个核苷酸的非编码 RNA 分子,参与 RNA 沉默和基因 (Gene) 表达的转录后调控。
![]() 图:RNA 通过转录 DNA 产生:因此,它是 DNA 中所含信息的直接拷贝。DNA 中含胸腺嘧啶(T,以蓝色标示),RNA 中含尿嘧啶(U,以紫色标示)。 许可:CC BY-NC 4.0 [@own_1_2024]
图:RNA 通过转录 DNA 产生:因此,它是 DNA 中所含信息的直接拷贝。DNA 中含胸腺嘧啶(T,以蓝色标示),RNA 中含尿嘧啶(U,以紫色标示)。 许可:CC BY-NC 4.0 [@own_1_2024]
在真核生物中,前体 mRNA 分子经过多种加工步骤以产生成熟的 mRNA 分子。为了稳定 mRNA,分子的 5' 端被加帽一个修饰的鸟嘌呤核苷酸(更具体地说,是 7-甲基鸟苷酸),3' 端被延伸一段长的腺嘌呤核苷酸链(称为多聚腺苷酸化)。此外,许多真核生物 mRNA 分子经历剪接 (Splicing)。在 RNA 剪接过程中,剪接体蛋白复合物去除内含子:mRNA 分子中在翻译过程中不使用的特定非编码部分,以创建成熟 mRNA。大多数内含子在其 5' 和 3' 端分别以 GU 和 AG 二核苷酸基序为特征。
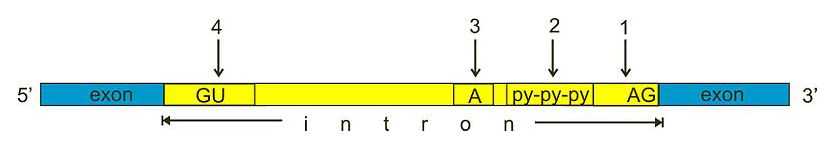 图:剪接过程中,从前体 mRNA 分子中去除内含子以创建成熟 mRNA。大多数内含子包含剪接体的识别序列,并产生特定的二级结构以提高剪接效率:(1) 3' 剪接位点,(2) 多嘧啶束,(3) 分支位点,(4) 5' 剪接位点。 许可:CC0 1.0 [@splicing_2011]
图:剪接过程中,从前体 mRNA 分子中去除内含子以创建成熟 mRNA。大多数内含子包含剪接体的识别序列,并产生特定的二级结构以提高剪接效率:(1) 3' 剪接位点,(2) 多嘧啶束,(3) 分支位点,(4) 5' 剪接位点。 许可:CC0 1.0 [@splicing_2011]
翻译¶
在蛋白质翻译 (Translation) 过程中,核糖体从信使 RNA (mRNA) 合成多肽。在此过程中,tRNA 将 RNA 上的信息解码为氨基酸,其中由三个核苷酸组成的密码子编码一个氨基酸的信息。
![]() 图:翻译过程,核糖体与 tRNA 分子通过反密码子"读取" mRNA 上的密码子,然后将其翻译为对应的氨基酸。这些氨基酸通过肽键连接形成多肽链。 许可:CC BY-NC 4.0 [@own_1_2024]
图:翻译过程,核糖体与 tRNA 分子通过反密码子"读取" mRNA 上的密码子,然后将其翻译为对应的氨基酸。这些氨基酸通过肽键连接形成多肽链。 许可:CC BY-NC 4.0 [@own_1_2024]
延伸阅读
转录和翻译的细节在原核生物和真核生物之间有所不同。你可以查阅 Biology 2e 的第 15 和第 16 章了解更多。
遗传密码¶
遗传密码展示了密码子与氨基酸之间的对应关系。由于 64 种可能的密码子编码 20 种不同的氨基酸,遗传密码是简并的,即大多数氨基酸由一个以上的密码子指定。因此,编码同一特定氨基酸的密码子可能在一个或两个位置上有所不同。你可以注意到,在遗传密码表中,编码相同氨基酸的密码子通常在第三个密码子位置上不同。由于密码的简并性,蛋白质序列可以从 DNA 或 RNA 序列推导出来,但反过来则不行。
有三个密码子不编码氨基酸,而是发出蛋白质序列结束的信号,称为**终止密码子**。此外,翻译通常以编码甲硫氨酸的起始密码子 AUG 开始。关于蛋白质信息如何在基因组中编码的更多信息,可以在基因组注释一节中找到。
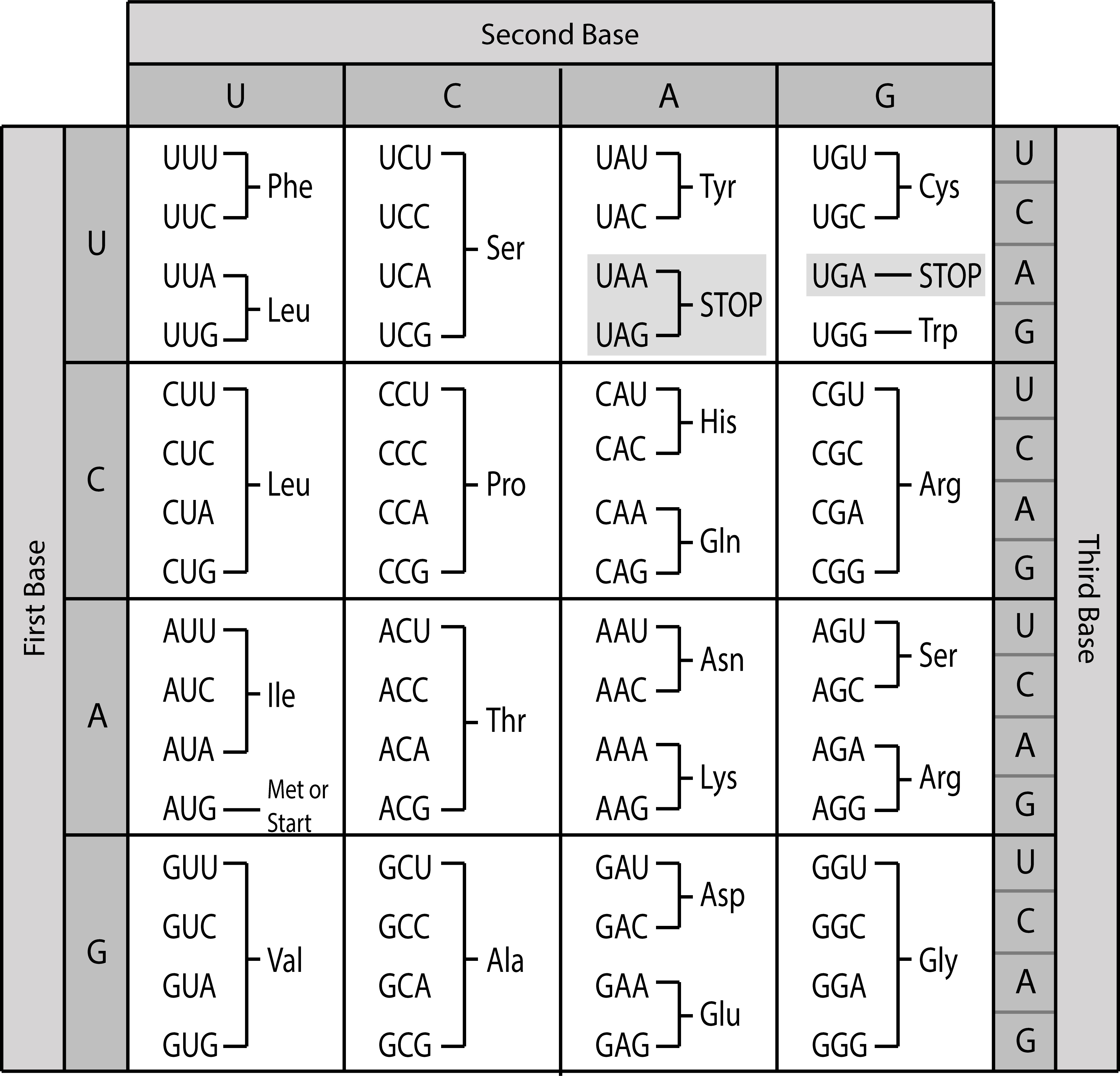 图:通用遗传密码。请注意,此密码存在例外,例如脊椎动物线粒体密码。 许可:CC BY 4.0 [@geneticcode_2018]
图:通用遗传密码。请注意,此密码存在例外,例如脊椎动物线粒体密码。 许可:CC BY 4.0 [@geneticcode_2018]
遗传密码速查表
通用遗传密码对于理解信息如何从基因流向蛋白质至关重要。尽管如此,你不需要记住它,可以随时查阅。在考试中,需要时也会提供。
分子生物学中心法则¶
根据分子生物学的中心法则,遗传信息的流动基本上是单向的:从 DNA 经 RNA 到蛋白质。然而,也有一些基因不编码蛋白质,其终产物是功能性 RNA。此外,可移动遗传元件和病毒可以编码逆转录酶(能够从 RNA 模板合成 DNA)或 RNA 依赖性 RNA 聚合酶(能够复制 RNA)。
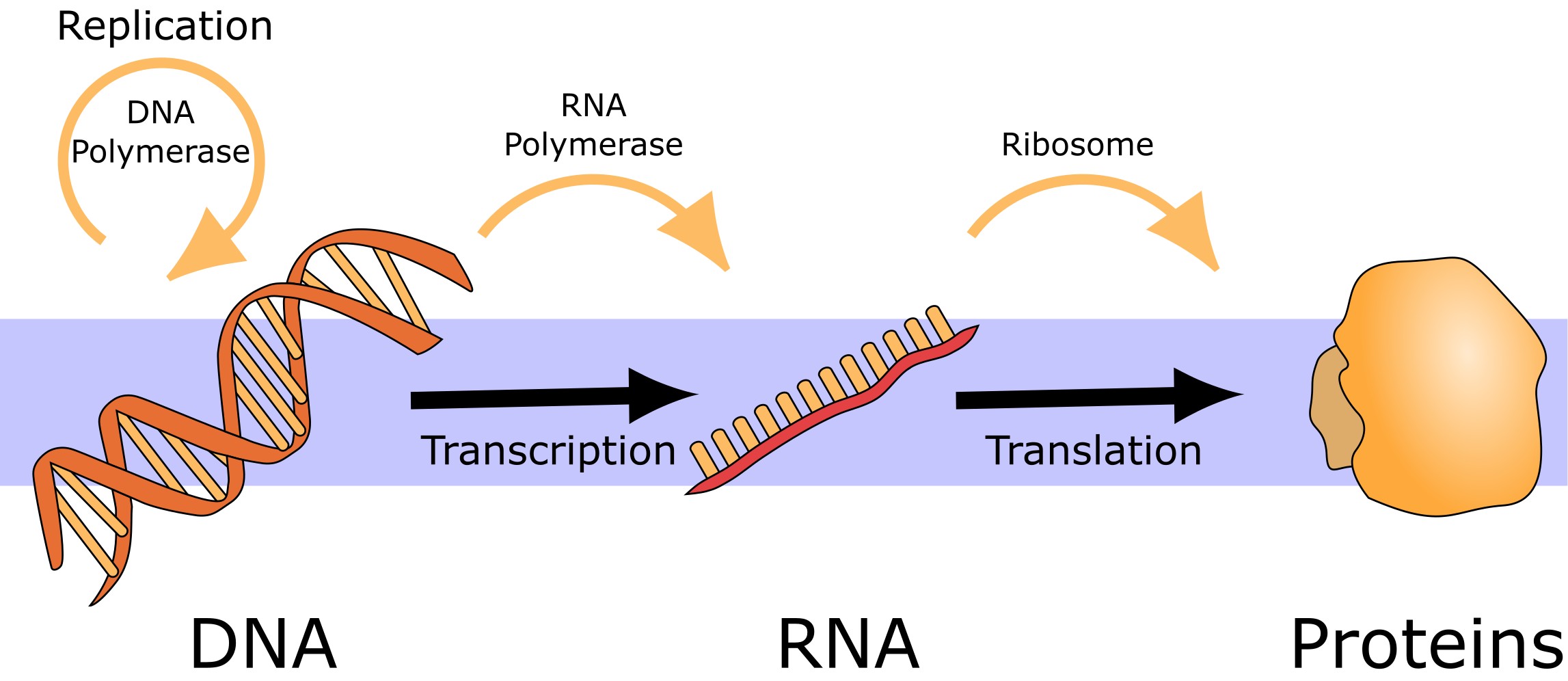 图:分子生物学中心法则。 许可:CC0 1.0 修改自 [@dogma_alt_2008]。
图:分子生物学中心法则。 许可:CC0 1.0 修改自 [@dogma_alt_2008]。
蛋白质¶
蛋白质是在体内发挥许多重要功能的大型复杂大分子。它们对细胞完成的大部分工作至关重要,并且是身体组织和器官的结构、功能和调节所必需的。蛋白质的基本构建块是氨基酸。
氨基酸¶
氨基酸包含一个中心碳原子(称为 α-碳,或 Cα)。α-碳连接着一个氨基(NH2)、一个羧基(COOH)和一个氢原子。此外,每种氨基酸都有一个特定的残基 (R) 基团。
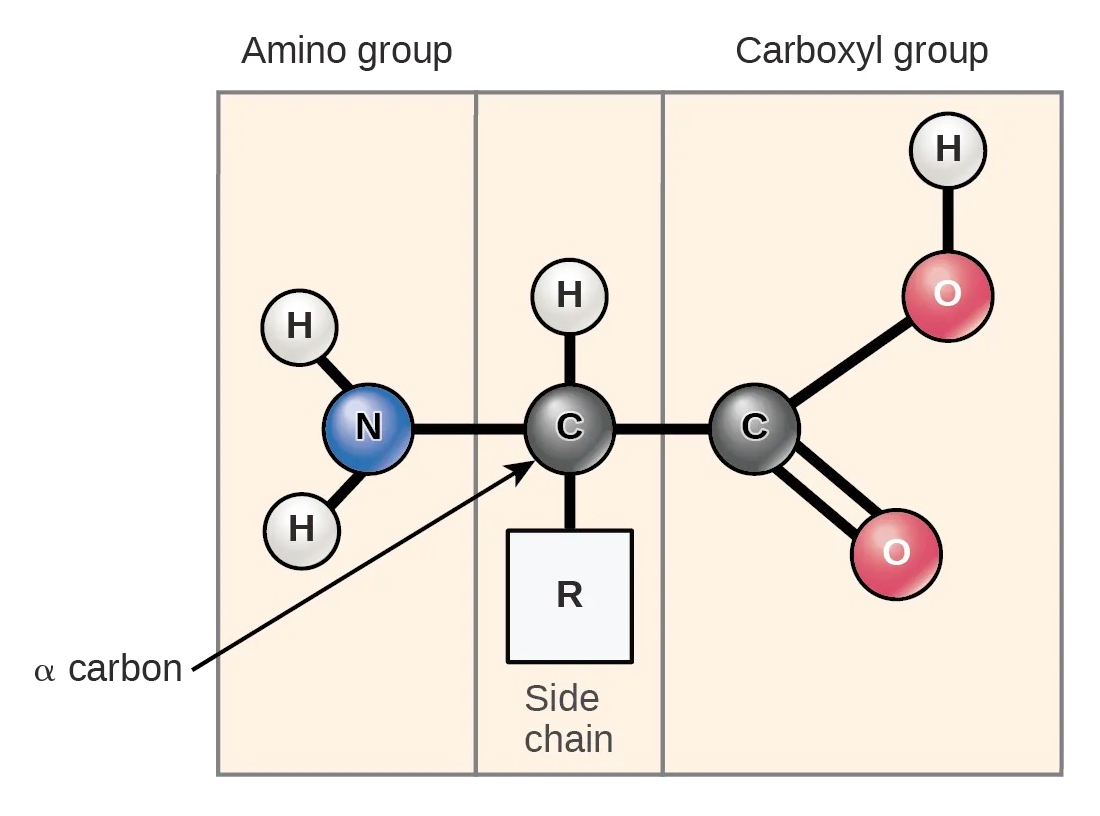 图:氨基酸的结构。四个元素连接到 α-碳:氨基、氢原子、羧基和侧链(R 基团)。 许可:CC BY 4.0 [@proteins_2018]
图:氨基酸的结构。四个元素连接到 α-碳:氨基、氢原子、羧基和侧链(R 基团)。 许可:CC BY 4.0 [@proteins_2018]
氨基酸的化学性质
氨基酸因化学性质而异,这些性质由它们的 R 基团决定。由于这些序列在生物信息学中无处不在,建立序列与结构和功能之间关系的直觉非常重要。因此,你应该熟记氨基酸、它们的一字母和三字母缩写,以及基本性质。
| 氨基酸 | 三字母缩写 | 一字母缩写 | 性质 |
|---|---|---|---|
| 精氨酸 (Arginine) | Arg | R | 正电荷 |
| 组氨酸 (Histidine) | His | H | 正电荷 |
| 赖氨酸 (Lysine) | Lys | K | 正电荷 |
| 天冬氨酸 (Aspartic acid) | Asp | D | 负电荷 |
| 谷氨酸 (Glutamic acid) | Glu | E | 负电荷 |
| 丝氨酸 (Serine) | Ser | S | 极性不带电荷 |
| 苏氨酸 (Threonine) | Thr | T | 极性不带电荷 |
| 天冬酰胺 (Asparagine) | Asn | N | 极性不带电荷 |
| 谷氨酰胺 (Glutamine) | Gln | Q | 极性不带电荷 |
| 丙氨酸 (Alanine) | Ala | A | 疏水性 |
| 缬氨酸 (Valine) | Val | V | 疏水性 |
| 异亮氨酸 (Isoleucine) | Ile | I | 疏水性 |
| 亮氨酸 (Leucine) | Leu | L | 疏水性 |
| 甲硫氨酸 (Methionine) | Met | M | 疏水性 |
| 苯丙氨酸 (Phenylalanine) | Phe | F | 疏水性和芳香性 |
| 酪氨酸 (Tyrosine) | Tyr | Y | 疏水性和芳香性 |
| 色氨酸 (Tryptophan) | Trp | W | 疏水性和芳香性 |
| 甘氨酸 (Glycine) | Gly | G | 特殊(侧链仅为 H) |
| 脯氨酸 (Proline) | Pro | P | 特殊(侧链与骨架氮原子连接) |
| 半胱氨酸 (Cysteine) | Cys | C | 特殊(形成二硫键) |
某些氨基酸具有非极性侧链,通常是**疏水性**的,即水分子无法与这些分子形成氢键。因此,它们通常可以在蛋白质内部与其他疏水性氨基酸一起被发现。**芳香族**氨基酸含有芳香环,常常稳定蛋白质的折叠结构。
相反,带电荷和极性氨基酸是**亲水性**的,即水分子可以与这些分子形成氢键。它们通常可以在蛋白质表面或与其他带相反电荷的氨基酸相互作用时在蛋白质内部被发现。**正电荷**氨基酸也称为碱性氨基酸,**负电荷**氨基酸也称为酸性氨基酸。
尽管氨基酸可以根据其性质分为这些类别,但有些氨基酸格外突出。最小的氨基酸是甘氨酸,由于其体积小,提供了很大的灵活性。相反,脯氨酸是一种侧链与骨架氮原子结合的氨基酸,使其非常刚性。最后,一个半胱氨酸氨基酸可以与另一个半胱氨酸形成二硫键。
蛋白质结构¶
蛋白质由一个或多个长链氨基酸折叠而成(每条链称为一个**多肽**)。蛋白质的三维结构也称为其**构象**。蛋白质构象分为四个层次来描述——从一级到四级结构。
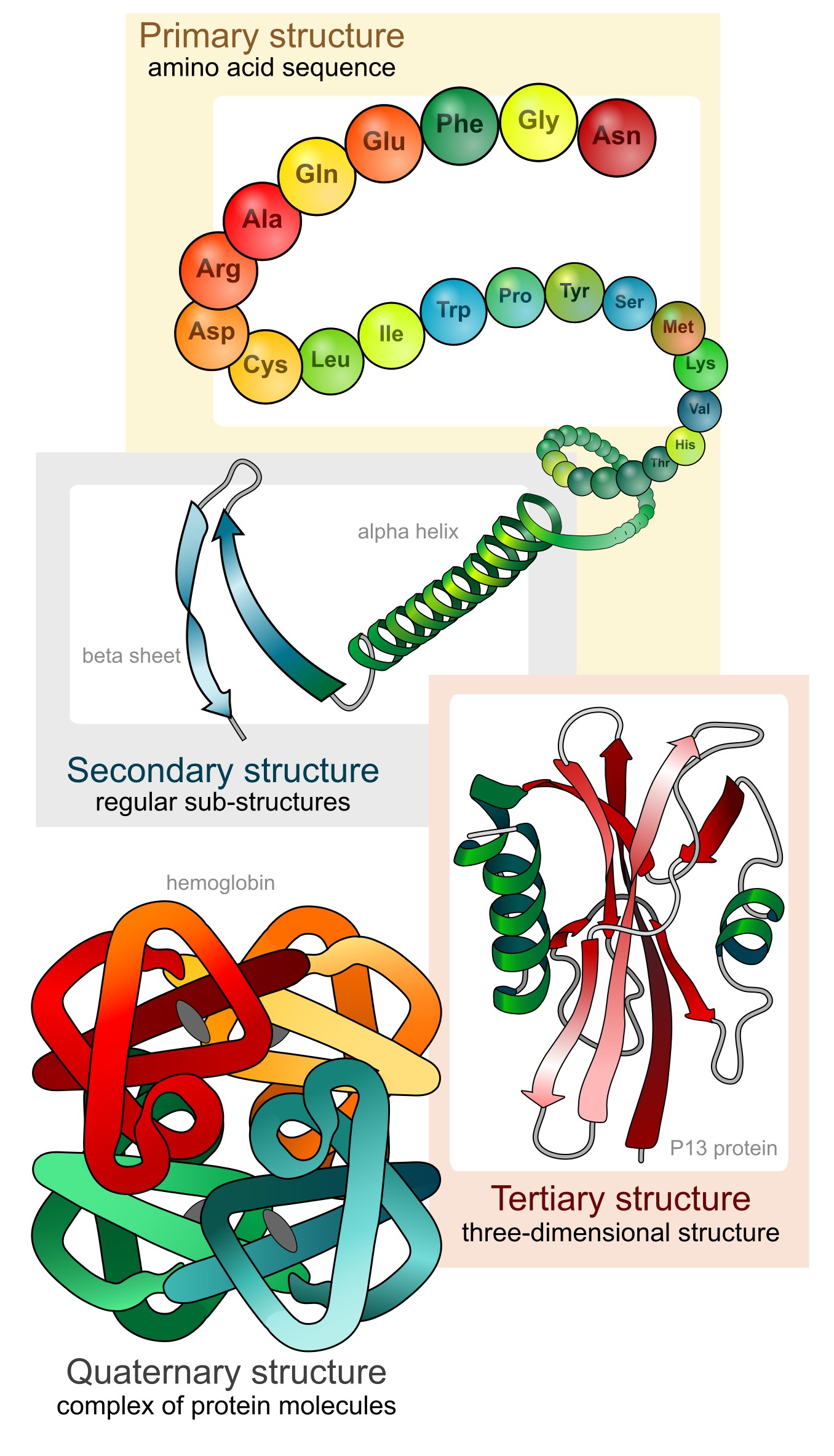 图:蛋白质结构的四个层次。 许可:CC0 1.0 [@struclevels_alt_2008]
图:蛋白质结构的四个层次。 许可:CC0 1.0 [@struclevels_alt_2008]
蛋白质的结构对其功能至关重要。例如,在酶中,活性位点必须具有正确的结构才能结合底物。其他蛋白质可能结合蛋白质(并影响其活性)或结合 DNA(并调节基因表达)。此外,有些蛋白质从细胞中分泌,或者可能在细胞膜内发挥作用。最后,蛋白质通常在蛋白质合成后(见翻译)被修饰,称为翻译后修饰。这些修饰可能对蛋白质功能非常重要。
一级结构¶
在蛋白质中,氨基酸通过共价键连接,称为肽键。肽键将一个氨基酸的羧基与下一个氨基酸的氨基连接起来。由肽键连接的氨基酸序列称为**一级结构**。蛋白质序列由编码该蛋白质的基因序列决定。沿着蛋白质的原子连续链也称为**骨架**,它由三个骨架原子(氮、Cα、碳)组成。
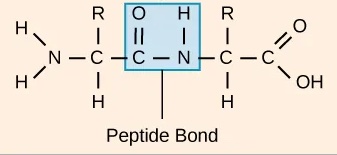 图:连接两个氨基酸的肽键。 许可:CC BY 4.0 [@proteins_2018]
图:连接两个氨基酸的肽键。 许可:CC BY 4.0 [@proteins_2018]
每个蛋白质在一端有一个自由的氨基,称为 N 端。另一端有一个自由的羧基,称为 C 端。
笔记 1.1:可能的多肽链数量
由于有 20 种不同的氨基酸,可以存在大量不同的多肽链,即对于长度为 n 的多肽有 20n 种。这些潜在序列中的大多数不会采用稳定的构象,因此只有极小一部分的可能性在自然界中存在。
二级结构¶
二级结构是蛋白质中由骨架原子之间的氢键稳定的局部构象。我们区分规则的螺旋(即 α-螺旋)和片层结构(即 β-折叠)以及不规则的转角。
**α-螺旋**由一个氨基酸 C 基团中的氧原子与沿链向后四个氨基酸的 N 基团中的氢之间的氢键稳定。每个螺旋圈有 3.6 个氨基酸残基,侧链从螺旋中伸出。
β-折叠片由 β-链组成,其中 R 基团从链的上下两侧延伸。链的方向由蛋白质的 N 端和 C 端决定,通常用指向 C 端的箭头表示。根据方向,链可以平行或反平行排列。
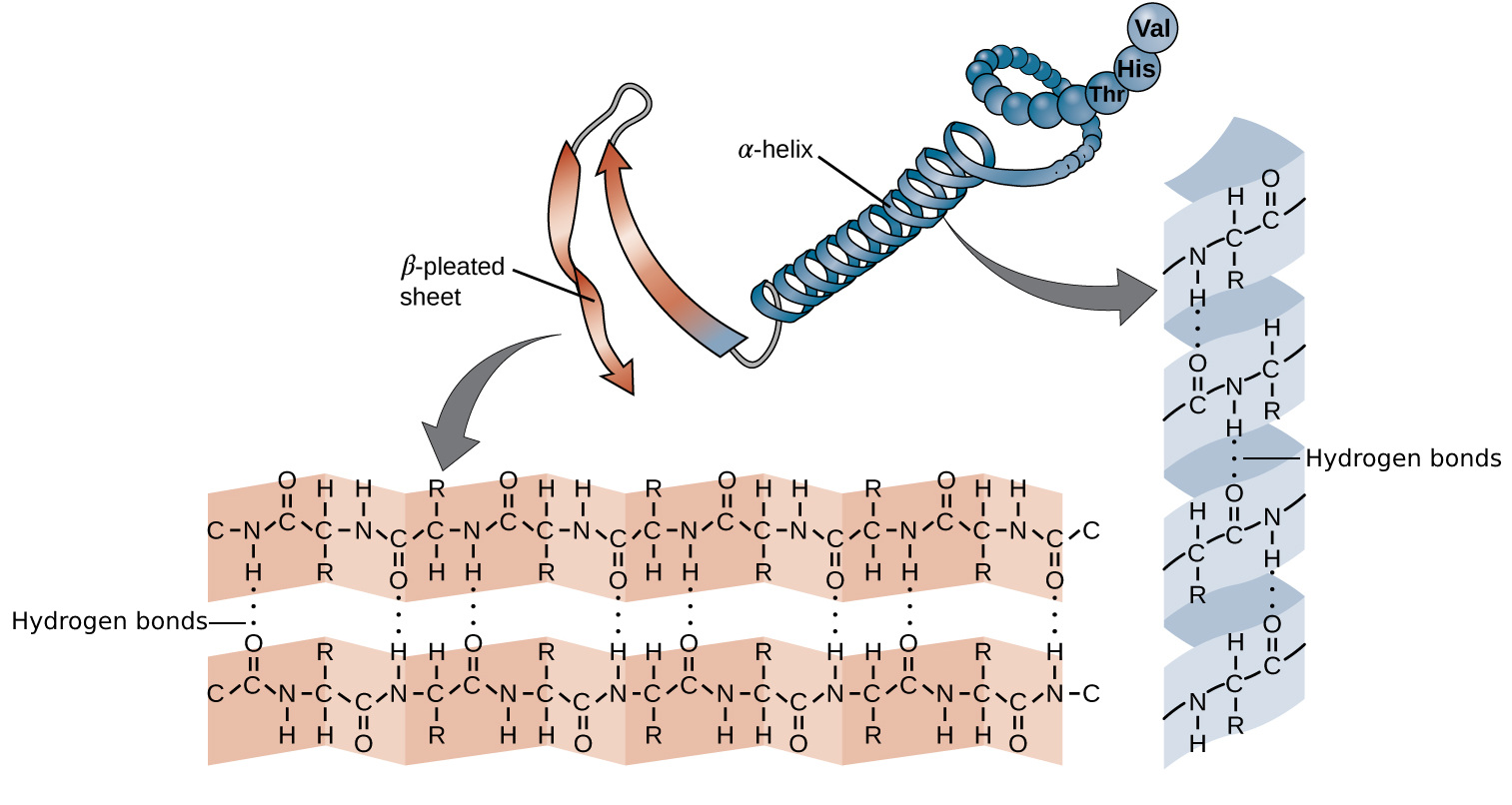 图:α-螺旋和 β-折叠由蛋白质骨架之间的氢键(虚线)稳定,即侧链不参与。氢键在一个氨基酸 C 基团中的氧原子与 N 基团中的氢之间形成。 许可:CC BY 4.0 修改自 [@secstructure_alt_nd]。
图:α-螺旋和 β-折叠由蛋白质骨架之间的氢键(虚线)稳定,即侧链不参与。氢键在一个氨基酸 C 基团中的氧原子与 N 基团中的氢之间形成。 许可:CC BY 4.0 修改自 [@secstructure_alt_nd]。
**转角**是由 1 到 5 个肽键距离内的氨基酸之间的氢键稳定的短二级结构元件。最常见的形式是 β-转角,连接反平行 β-链。
笔记 1.2:二级结构的氨基酸偏好
虽然二级结构元件由骨架之间的氢键形成,但某些氨基酸在二级结构中受到偏好,而另一些则不受偏好。例如,甲硫氨酸、丙氨酸、亮氨酸和谷氨酸在 α-螺旋中受偏好,而脯氨酸、甘氨酸和酪氨酸则不受偏好。此外,与 α-螺旋相比,缬氨酸、异亮氨酸、酪氨酸、半胱氨酸、色氨酸、苯丙氨酸和苏氨酸在 β-折叠中出现频率更高。在转角中,甘氨酸、天冬酰胺、脯氨酸和丝氨酸受到偏好。这些偏好被用于预测蛋白质中的二级结构元件(见第 4 章)。
肽键非常刚硬且呈平面状,即它不能旋转来形成蛋白质结构元件。然而,N-Cα 和 Cα-C 键可以自由旋转,仅受 R 基团大小和性质的限制。多肽骨架的三维形状因此由两个**扭转角**决定:N 和 Cα 之间的 phi (φ) 以及 Cα 和 C 之间的 psi (ψ)。虽然 φ 和 ψ 原则上可以旋转,但空间位阻阻止了某些角度组合,即 R 基团的体积限制了可能的构象。因此,某些 φ 和 ψ 的组合更受偏好。我们可以在所谓的**Ramachandran 图**中绘制蛋白质中 φ 和 ψ 的组合。
规则的二级结构元件(α-螺旋和 β-折叠)包含具有相似 (φ, ψ) 值的连续氨基酸。这些区域在 Ramachandran 图中通常高度集中。因此,Ramachandran 图可用于评估预测蛋白质结构的合理性。
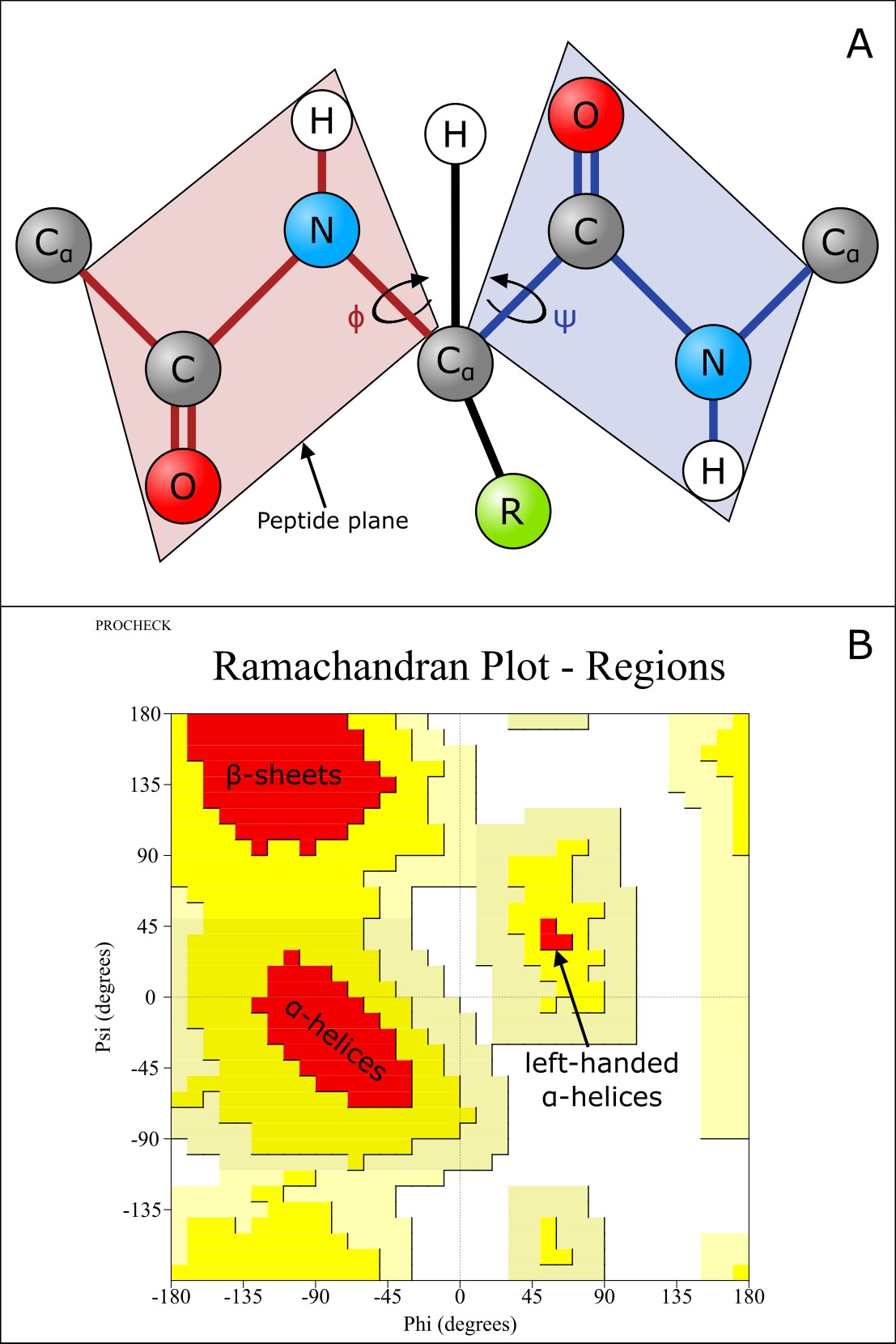 图:A)多肽链的 φ 和 ψ 扭转角。许可:CC BY-NC 4.0 [@own_1_2024]。B)典型的 Ramachandran 图。红色标记区域没有空间位阻,黄色区域代表具有空间位阻的构象,浅黄色区域代表通常空间上不利的构象,白色区域没有允许的构象。Ramachandran 图修改自 PROCHECK [@procheck_1993]。
图:A)多肽链的 φ 和 ψ 扭转角。许可:CC BY-NC 4.0 [@own_1_2024]。B)典型的 Ramachandran 图。红色标记区域没有空间位阻,黄色区域代表具有空间位阻的构象,浅黄色区域代表通常空间上不利的构象,白色区域没有允许的构象。Ramachandran 图修改自 PROCHECK [@procheck_1993]。
延伸阅读
关于 φ 和 ψ 的动画演示,请参阅 YouTube 视频。
三级结构¶
蛋白质的三级结构描述了整个多肽链的完全折叠。与二级结构不同,蛋白质的三级结构涉及氨基酸侧链之间的相互作用,这些相互作用可以在近距离和远距离发生。因此,氨基酸的化学性质对三级结构非常重要。不同类型的相互作用稳定了三级结构:
- 涉及极性氨基酸的氢键。
- 正负电荷氨基酸之间的离子键。
- 倾向于位于蛋白质内部的疏水性 R 基团,通过疏水相互作用稳定。
- 二硫键(即半胱氨酸之间的共价键)。
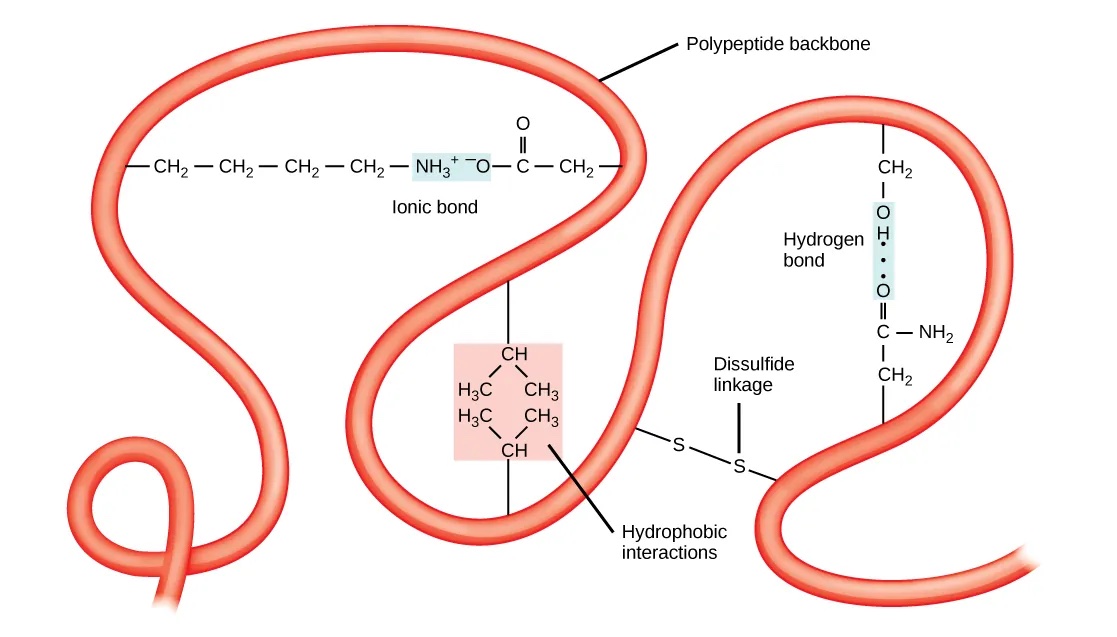 图:稳定蛋白质三级结构的化学相互作用。 许可:CC BY 4.0 [@proteins_2018]
图:稳定蛋白质三级结构的化学相互作用。 许可:CC BY 4.0 [@proteins_2018]
笔记 1.3:变性
稳定蛋白质结构的非共价键在高温下会被破坏。因此,大多数蛋白质在约 60°C 以上会展开。这个过程称为变性,通常是不可逆的。当蛋白质变性时,它们会失去功能。
在研究许多不同的蛋白质结构时,可以观察到各种反复出现的子结构。这些所谓的**结构域 (Domains)** 是蛋白质中不同的功能性和/或结构单元,通常长度为 50 到 350 个氨基酸。通常,一个结构域负责特定的功能或相互作用,为蛋白质的整体功能做出贡献。一个结构域可以与其它结构域在不同上下文中存在。在多结构域蛋白质中,每个结构域独立于其他结构域折叠。
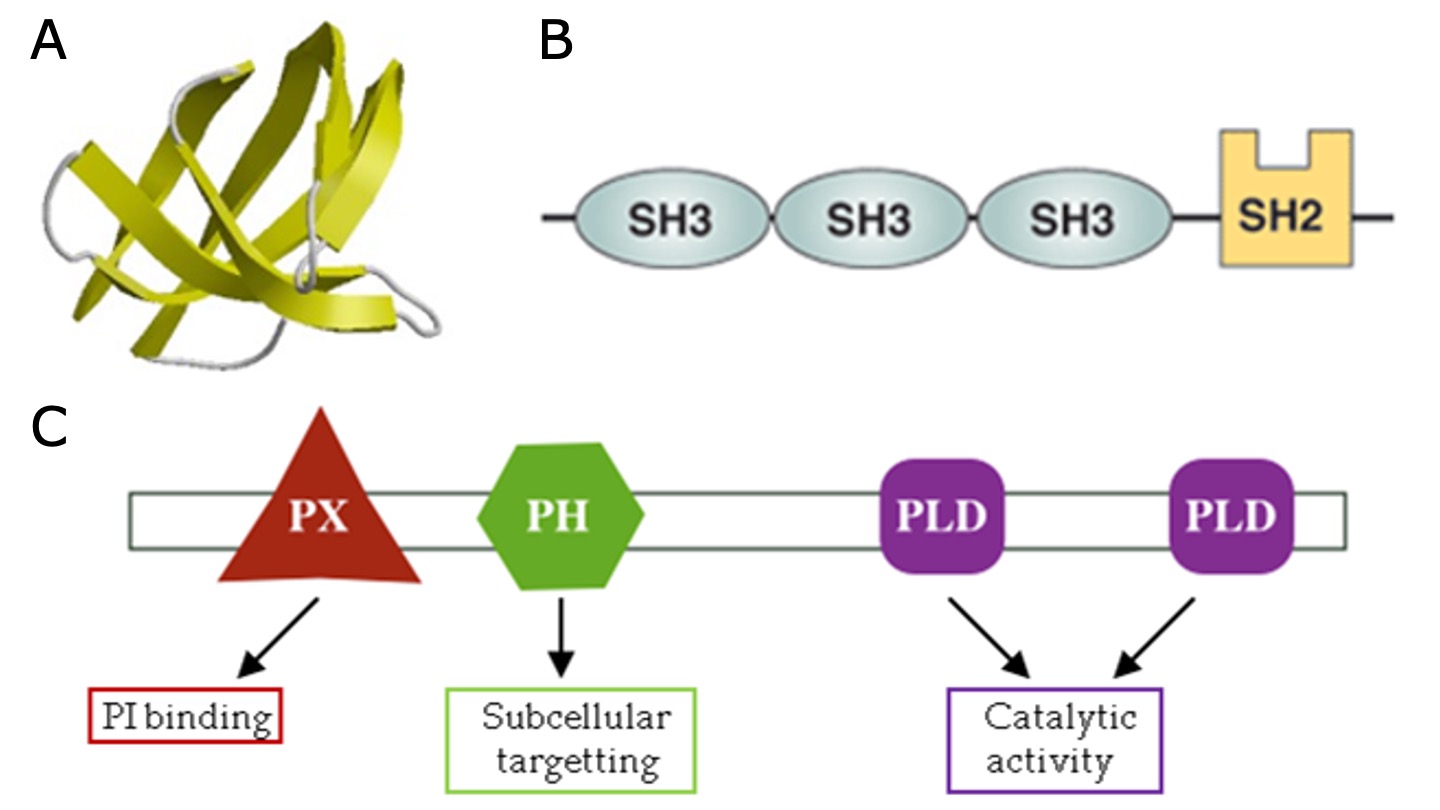 图:A)参与蛋白质-蛋白质相互作用的 Src 同源 3 (SH3) 结构域示例。SH3 结构域出现在具有不同功能的多种蛋白质中。B)细胞质蛋白 Nck 包含多个 SH3 结构域。C)磷脂酶 D1 的结构域组成,它具有多个为其整体功能做出贡献的功能结构域。 许可:CC BY 4.0 [@domains_2023]
图:A)参与蛋白质-蛋白质相互作用的 Src 同源 3 (SH3) 结构域示例。SH3 结构域出现在具有不同功能的多种蛋白质中。B)细胞质蛋白 Nck 包含多个 SH3 结构域。C)磷脂酶 D1 的结构域组成,它具有多个为其整体功能做出贡献的功能结构域。 许可:CC BY 4.0 [@domains_2023]
四级结构¶
最后,单独折叠的多肽可以相互作用形成**蛋白质复合体**,也称为四级结构。四级结构由与三级结构相同类型的相互作用稳定。区别在于所涉及的氨基酸属于不同的多肽。
许多功能性蛋白质由多个亚基组成,也称为**寡聚体**。亚基可以来自相同的蛋白质序列(称为同源寡聚体)或来自不同的序列(称为异源寡聚体)。由两个亚基组成的蛋白质也称为二聚体。
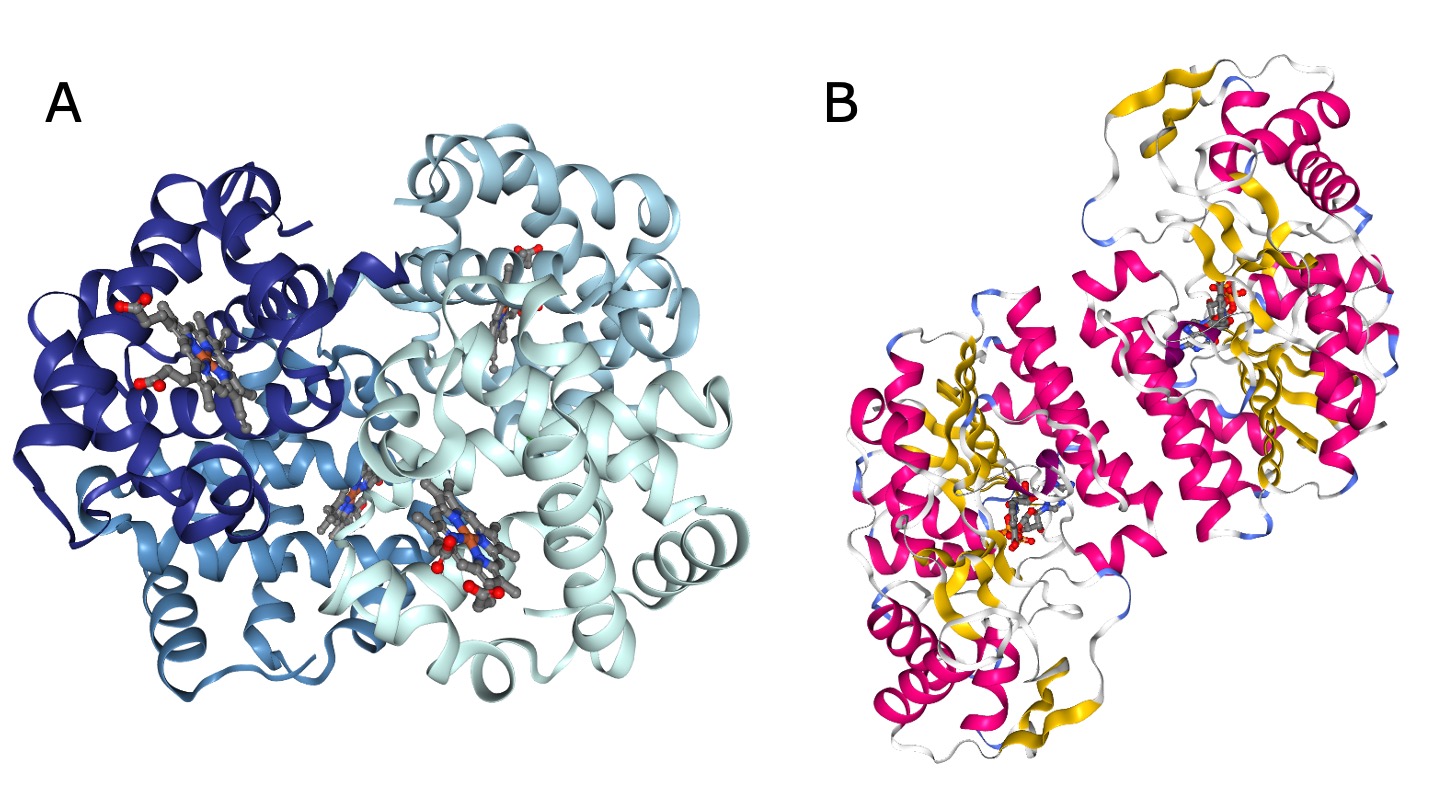 图:寡聚体示例。A)肌红蛋白,四个亚基的异源寡聚体(PDB 结构 1HV4,按链着色)[@rcsb_2000, @oligomers_a_2001, @ngl_2018]。B)UDP-半乳糖 4-差向异构酶,同源二聚体(PDB 结构 1EK5,按二级结构着色)[@rcsb_2000, @oligomers_b_2000, @ngl_2018]。
图:寡聚体示例。A)肌红蛋白,四个亚基的异源寡聚体(PDB 结构 1HV4,按链着色)[@rcsb_2000, @oligomers_a_2001, @ngl_2018]。B)UDP-半乳糖 4-差向异构酶,同源二聚体(PDB 结构 1EK5,按二级结构着色)[@rcsb_2000, @oligomers_b_2000, @ngl_2018]。
替换¶
基因序列中的突变可能导致蛋白质一级结构的变化,例如用一个氨基酸替换为另一个不同的氨基酸。通常,这种替换仍然导致高度相似的蛋白质结构,执行相似甚至相同的功能,特别是当被替换的氨基酸具有相似的化学性质时。然而,单个氨基酸替换也可能产生严重的后果。一个著名的例子是镰状细胞贫血症,其中血红蛋白 β 中谷氨酸到缬氨酸的替换导致结构变化,进而引起红细胞变形。
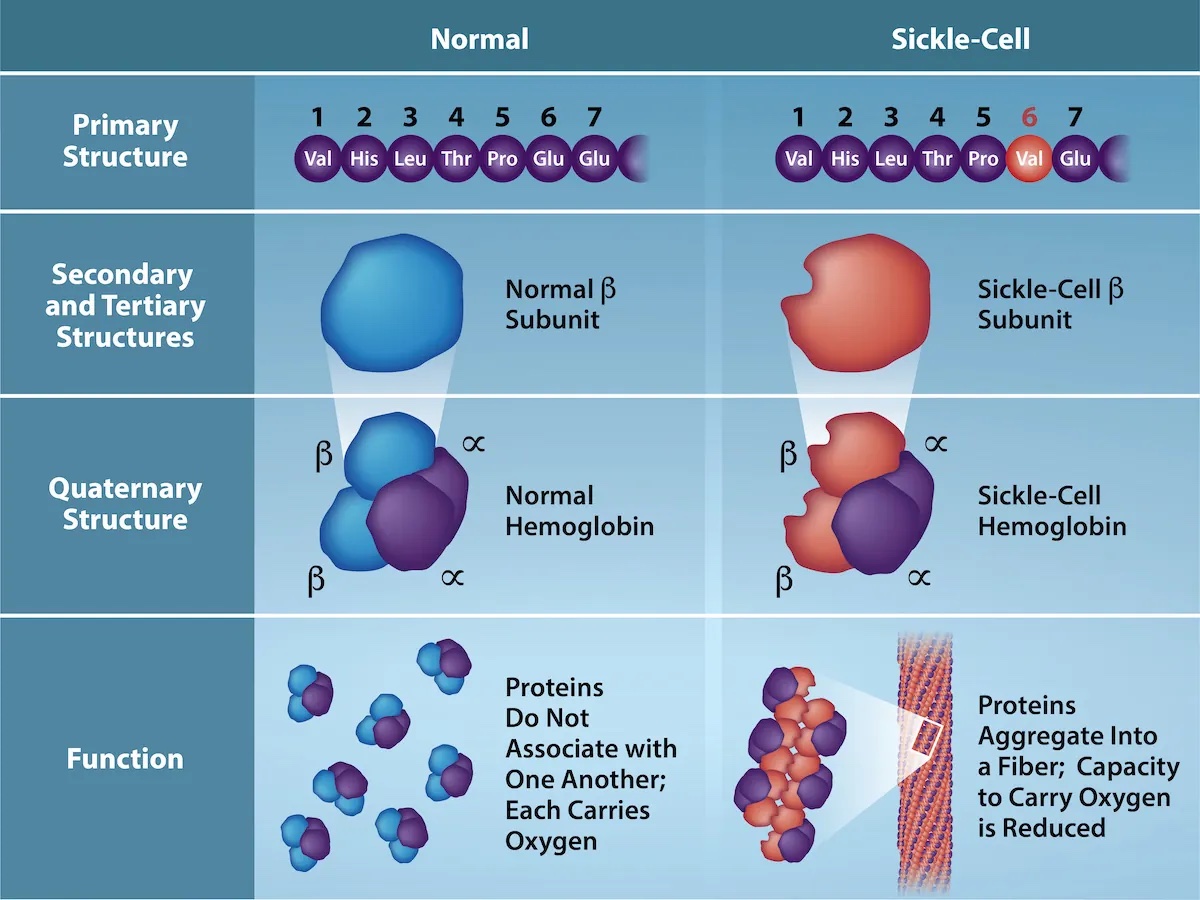 图:血红蛋白 β 中替换导致镰状细胞贫血症的后果。 来源:Rao, A., Tag, A. Ryan, K. and Fletcher, S. Department of Biology, Texas A&M University。
图:血红蛋白 β 中替换导致镰状细胞贫血症的后果。 来源:Rao, A., Tag, A. Ryan, K. and Fletcher, S. Department of Biology, Texas A&M University。
可视化¶
查看蛋白质分子结构有很多风格。有些风格侧重于详细的化学结构,其他风格则针对蛋白质表面。
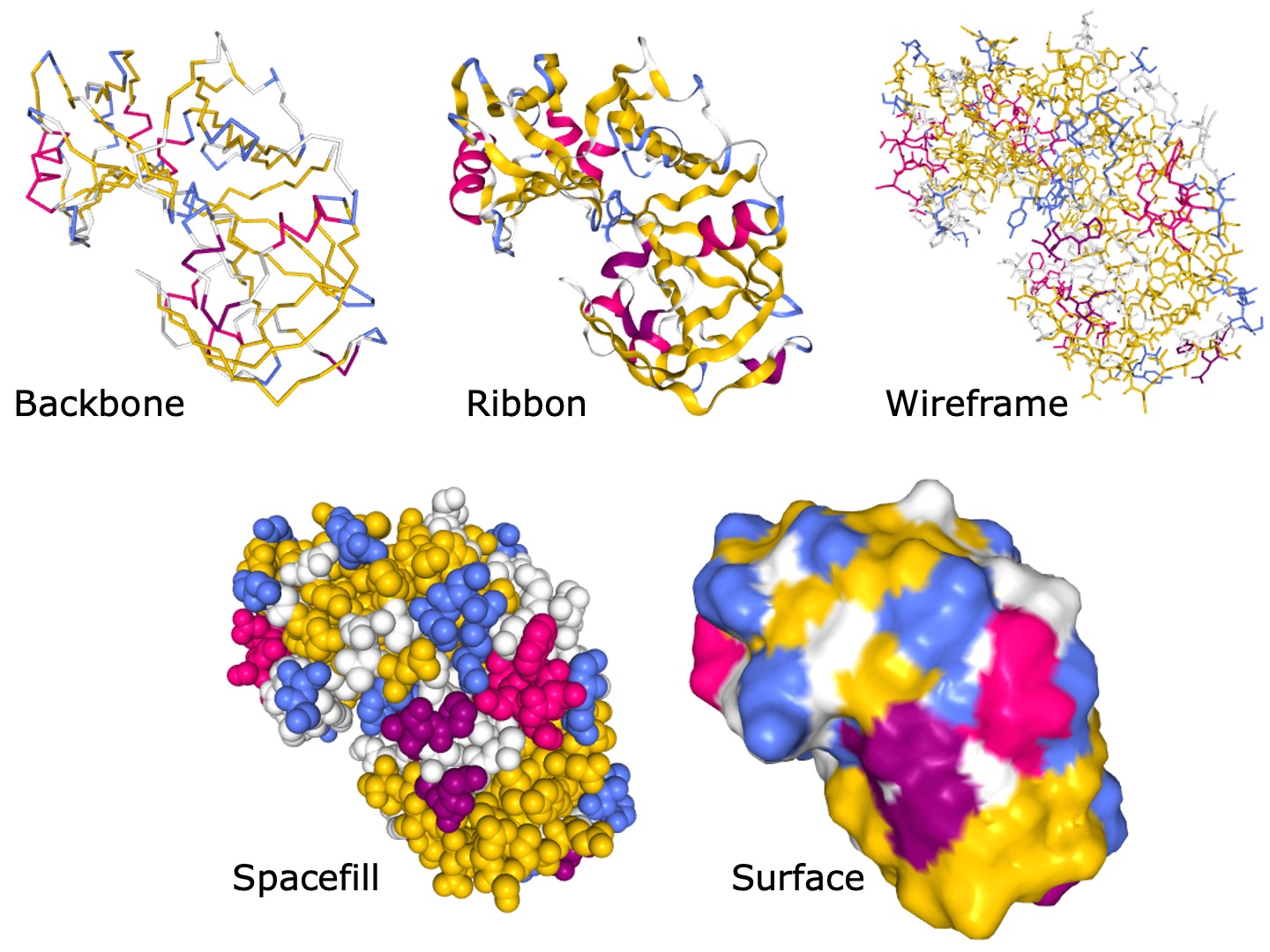 图:使用 NGL 生成的 PDB 结构 5PEP 的不同表示方式。[@rcsb_2000, @protrep_1990, @ngl_2018]
图:使用 NGL 生成的 PDB 结构 5PEP 的不同表示方式。[@rcsb_2000, @protrep_1990, @ngl_2018]
延伸阅读
本节中的大部分图片来自 OpenStax,你可以在那里找到更多关于蛋白质的信息。
基因组注释¶
基因组注释 (Annotation) 是解读生物体 DNA 中编码了什么信息的过程。对于已知基因组序列的生物体来说,这是一项持续的工作。更重要的是,基因组注释是从新测序的基因组中获取生物学见解的关键步骤。鉴于任何基因组的大小,自动化程序被用于识别各种基因组元件,如基因、调控区域、转座子元件或其他非编码元件。这些生物信息学程序通常专注于识别一种类型的元件,因此一个完整的基因组注释项目可以被视为各种程序的流水线 (Pipeline)。以下小节描述了基因组注释中最常见的步骤。
笔记 1.4:比对算法
基因组注释过程中的多个步骤利用可以搜索或比对生物序列的算法,例如 BLAST 算法。第 2 章 更详细地介绍了序列比对和搜索。目前,你只需要知道这些算法可以快速搜索非常大的生物序列集合来识别看起来相似的序列(我们所说的"相似"的准确含义也是第 2 章 的一部分)。
重复序列屏蔽¶
重复序列屏蔽涉及基因组中重复序列的识别和屏蔽(隐藏)。这是注释大多数基因组的重要第一步,因为重复序列可能给基因组注释带来重大挑战。屏蔽重复序列通常可以改善:
- 准确性:重复元件可能被错误地注释为基因或其他功能元件,导致对基因组的预测和解释不准确。
- 计算效率:识别和处理重复序列在计算上可能非常密集。然而,屏蔽这些重复区域减少了所有下游分析的计算时间。
- 生物学相关性:重复序列通常不参与编码感兴趣的蛋白质。因此,关注非重复区域是理解驱动生物学过程的基因和调控元件的明智选择。
大多数重复屏蔽工作流程首先编译(或使用预编译的)"重复文库":以前已经表征的已知重复元件的集合。随后,使用各种计算算法(如特定配置版本的 BLAST 或 RepeatMasker)将待注释的基因组与此重复文库进行比较。当找到匹配时,基因组中相应的区域被"屏蔽"或注释为重复元件。这意味着这些区域被排除在进一步分析之外或标记为重复序列。
基因预测¶
在原核生物和真核生物基因组中寻找蛋白编码基因的过程有所不同。在两种情况下,目标都是找到开放阅读框 (ORF):编码蛋白质的连续核苷酸片段。更具体地说,ORF 以起始密码子开始,以终止密码子结束,其长度是三的倍数(参考遗传密码)。由于 RNA 剪接在原核生物基因组中几乎不存在,原核生物的 ORF 可以直接在基因组 DNA 中找到。因此,简单地枚举基因组中所有可能的 ORF 是原核生物基因组注释中的常见步骤。相反,真核生物基因组中的 ORF 在成熟的 mRNA 上找到。因此,所有真核生物基因预测方法都考虑了剪接,从而大大增加了其计算复杂性。原核生物和真核生物基因预测通常都可以分为基于证据的预测或从头 (ab initio) 预测,下面都将解释。
基于证据的预测¶
这种数据驱动的方法使用现有的和新生成的数据来获取基因组哪些区域编码基因的线索。根据数据类型,这些预测具有或多或少的预测能力。一些常用的证据类型包括:
- RNA 测序数据:关于基因组哪些区域被转录的最直接证据。因此,RNA 测序(通常缩写为 RNA-seq)"读段"通常为识别真核生物中的剪接位点提供最佳证据。注意,并非所有转录的 RNA 都会被翻译为蛋白质,因此并非所有 RNA 测序读段都是蛋白编码基因的证据。区分编码蛋白质的 RNA 和非编码 RNA 并不总是容易的。
- 同源性证据:将已知基因(来自其他生物体)的 DNA 或蛋白质序列与基因组进行比对是寻找基因组编码区域的有价值证据。由于遗传密码的冗余性,将蛋白质序列与基因组比对时正确识别剪接位点并不容易。来自亲缘关系较近生物的同源性证据比来自远缘生物的证据产生更高质量的预测。
- 全基因组比对:这种方法使用亲缘关系较近生物的已注释基因组来直接识别新基因组中的编码区域。例如:小鼠和人类基因组的全基因组比对揭示小鼠 2 号染色体的大部分区域与人类 20 号染色体同源。比对过程产生小鼠和人类基因组坐标之间的直接一对一映射,因此注释坐标可以在基因组之间转移。
从头预测¶
Ab initio(拉丁语):从第一性原理出发,从头开始
这些方法依赖统计学从已知注释基因组中学习预测模型。存在多种形式的从头模型,虽然实现细节不同,但大多数遵循相似的推理方式。目前,我们将坚持高层次描述。所有从头模型扫描 DNA 序列并在每个位置给出特定类型注释的分数。此外,它们通常考虑特定位置的基因组上下文。例如,当接下来的两个观察到的核苷酸是 T 和 G 时,产生 ATG 起始密码子甲硫氨酸,核苷酸 A 的蛋白编码注释的概率很高。此外,大多数方法还考虑基因组上下文的_预测注释_。例如:如果我们还可以预测一个框内终止密码子,ATG 实际编码起始密码子的概率要高得多。在真核生物基因组预测中,这些模型变得相当复杂,因为它们必须在所有三个阅读框中包含剪接位点。模型究竟如何决定给哪个核苷酸什么注释分数是模型架构和参数化的一部分。在所有情况下,模型参数被选择来准确再现已知的基因组注释。如果使用足够的数据来学习模型参数,则假定这些模型可以用于预测新基因组序列上的注释。与基于同源性的预测一样,这种基于模型的方法对于亲缘关系较近的生物体效果最好。过去,几乎所有从头预测方法都表述为隐马尔可夫模型 (HMM)(见笔记 1.5)。实现基于 HMM 从头预测的工具示例包括 SNAP、GeneMark 和 Augustus。随着更多高质量数据(基因组序列和伴随注释)的可用性,基于深度学习和生成式 AI 的方法已被证明在性能上经常优于基于 HMM 的方法。
笔记 1.5:隐马尔可夫模型 (HMM)
隐马尔可夫模型 (HMM) 是一种分析序列的统计工具。它们在生物信息学中被广泛用于研究 DNA 或蛋白质序列等生物学数据。
完整的技术描述超出了本书的范围。取而代之,我们给出一个简化的介绍。
HMM 在一系列观测值上预测隐藏的标签。例如,在基因组注释中,基因组被观察为核苷酸序列,而编码和非编码区域是隐藏的标签。"隐藏"一词指的是这些未观察到的标签。"马尔可夫"一词指的是关于连续标签之间依赖性的统计假设,这使得计算高效。
形式上,HMM 具有:
- 隐藏状态:未观察到的标签(例如编码或非编码)。
- 发射概率:在给定隐藏状态下观察到一个符号(例如核苷酸)的可能性。
- 转移概率:从一个隐藏状态移动到另一个隐藏状态的可能性。
这些元素共同允许回答以下问题:
给定我当前的观察(例如核苷酸)和前一位置的标签,现在最可能的标签是什么?
在基因组注释中,这可能变成:
鉴于我看到一个终止密码子,而前一个标签是编码序列,当前标签是什么?
最可能的答案:非编码。
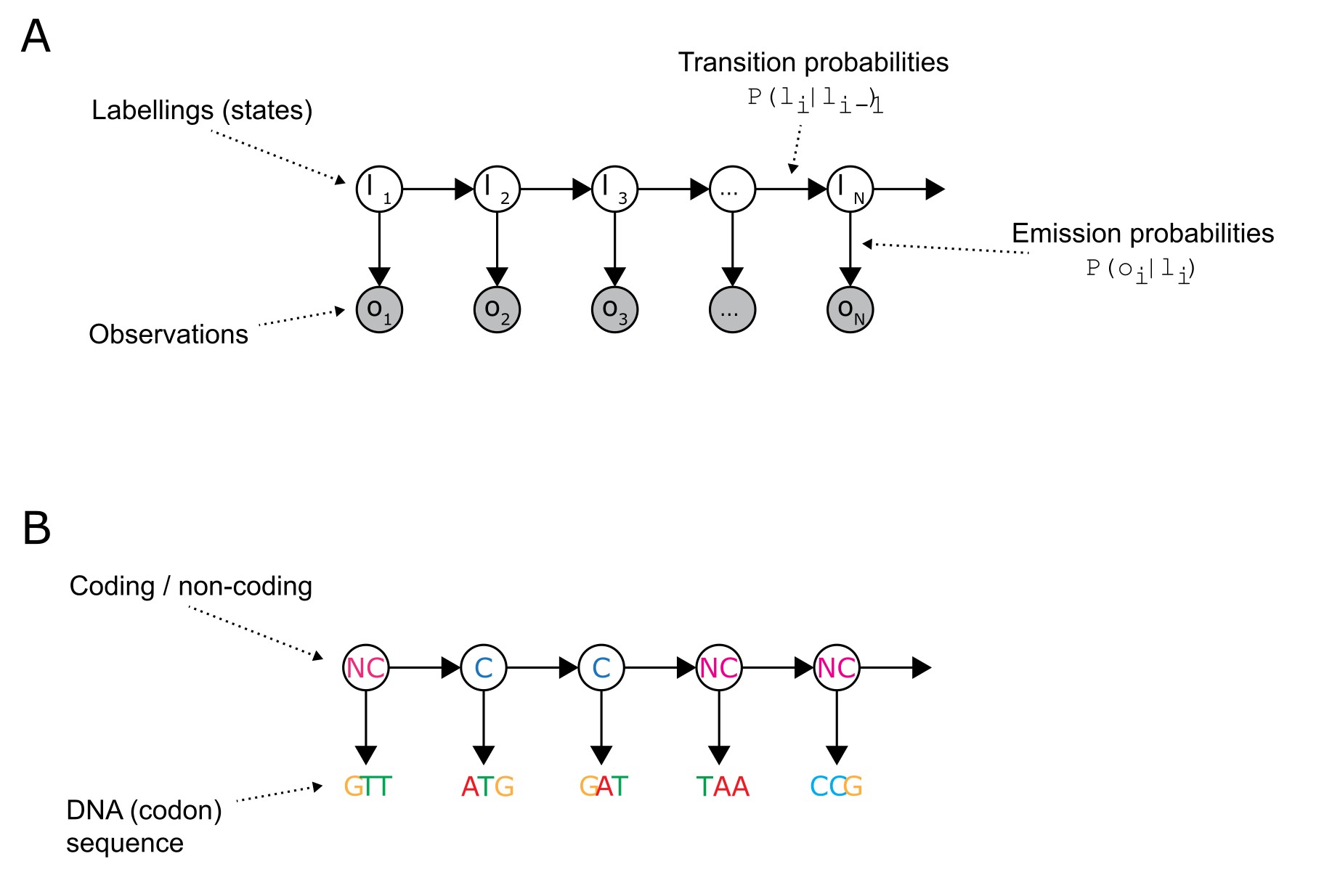 图:A:一般隐马尔可夫模型的图形表示。阴影圆表示观测值,白色圆表示未观察到的标签(隐藏状态)。黑色箭头表示隐藏状态之间的转移概率,以及从隐藏状态发射观测值的概率。注意观测值之间没有箭头!这是使计算高效的 HMM 特性之一。B:一个(简化的)HMM 变体,将 DNA 密码子序列标记为编码或非编码。实际基因预测 HMM 使用更复杂的结构,具有更多隐藏状态和 DNA 的六框表示。 许可:CC BY-NC 4.0 [@own_1_2024]
图:A:一般隐马尔可夫模型的图形表示。阴影圆表示观测值,白色圆表示未观察到的标签(隐藏状态)。黑色箭头表示隐藏状态之间的转移概率,以及从隐藏状态发射观测值的概率。注意观测值之间没有箭头!这是使计算高效的 HMM 特性之一。B:一个(简化的)HMM 变体,将 DNA 密码子序列标记为编码或非编码。实际基因预测 HMM 使用更复杂的结构,具有更多隐藏状态和 DNA 的六框表示。 许可:CC BY-NC 4.0 [@own_1_2024]
证据/预测整合¶
从前面的章节中,现在已经清楚有多种方法可以预测基因组中的基因是什么样的。由于这些不同方法几乎从不完全一致,基因组注释的最后一步是证据和预测整合。通常使用加权共识方法:每个单独的证据来源被赋予一个权重,表示它应该对最终决定产生多大影响,然后多数投票决定注释应该是什么样的。通常,RNA-seq 证据获得高权重,各种形式的同源性证据可以根据它们与目标基因组的亲缘关系进行加权。
功能注释¶
到目前为止,基因组注释过程中描述的所有步骤都涉及基因在结构层面的样子。为了获得生物学见解,下一步是将功能注释分配给预测的基因。这一功能注释步骤包括使用各种序列比对和搜索工具来查找具有已知功能/描述的序列,并将已知基因的信息转移到预测基因上。通常使用多种高质量已知功能数据库,这些数据库将在本章的下一节中更详细地描述。在第 2 章中,我们将学习如何高效搜索这些数据库。
笔记 1.6:可视化基因结构
基因模型:基因的基因组结构(通常称为基因"模型")通常用一组具有预定义含义的线和矩形来可视化。
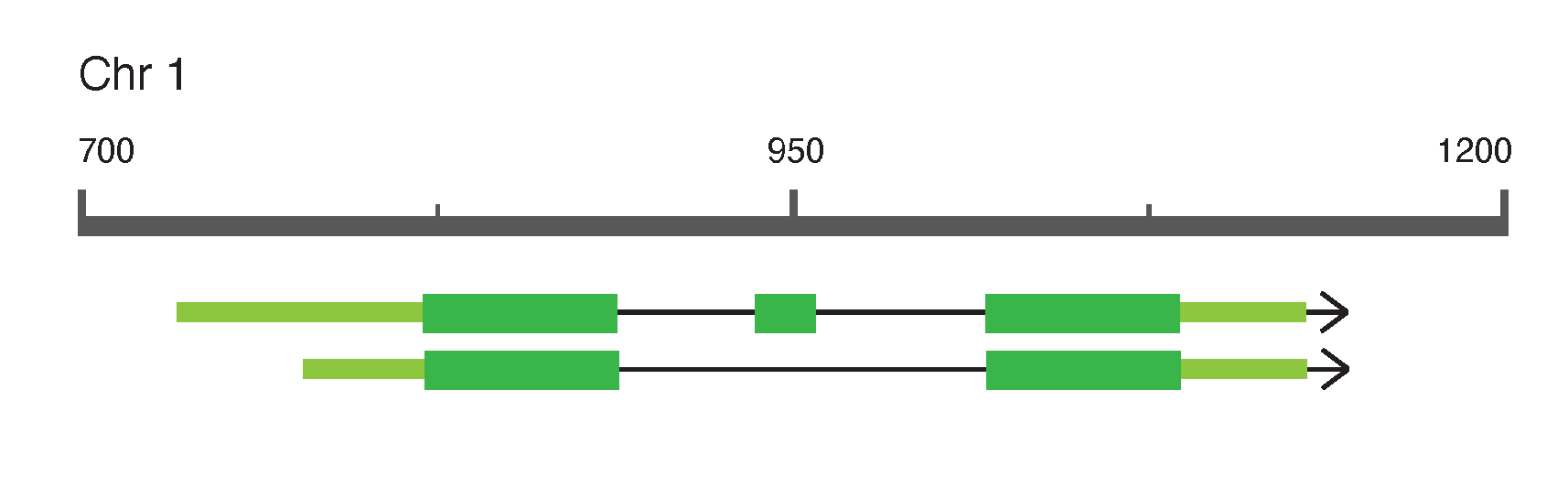 图:一个基因模型示例。可以识别各种可视化约定:方框表示被转录的基因组区域。方框是外显子 (Exon),方框之间的线是内含子 (Intron)。窄方框(有时颜色较浅)是非转录区域 (UTR),宽方框(有时颜色较深)是编码序列区域 (CDS)。箭头表示转录方向。在此示例中展示了 1 号染色体上的一个具有两种剪接变体的基因,其中第一个变体的 5' UTR 稍长,在第一个和最后一个外显子之间还有一个额外的 CDS 外显子。 许可:CC BY-NC 4.0 [@own_1_2024]
图:一个基因模型示例。可以识别各种可视化约定:方框表示被转录的基因组区域。方框是外显子 (Exon),方框之间的线是内含子 (Intron)。窄方框(有时颜色较浅)是非转录区域 (UTR),宽方框(有时颜色较深)是编码序列区域 (CDS)。箭头表示转录方向。在此示例中展示了 1 号染色体上的一个具有两种剪接变体的基因,其中第一个变体的 5' UTR 稍长,在第一个和最后一个外显子之间还有一个额外的 CDS 外显子。 许可:CC BY-NC 4.0 [@own_1_2024]
基因组浏览器 (Genome browser) 便于在基因组序列上交互式可视化注释和证据比对。存在各种实现,但所有基因组浏览器通常提供染色体的线性视图,可以滚动和缩放。此外,可以经常切换各种注释"轨道",以显示例如已知的基因结构、RNA 测序比对或同源蛋白质序列比对。大多数可视化元素可以点击以打开包含附加信息的弹出窗口。
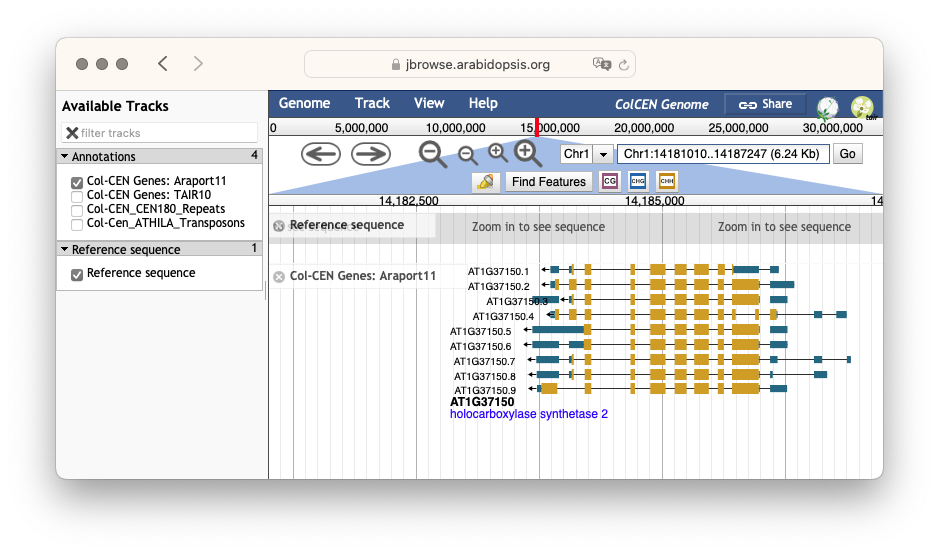 图:JBrowse 基因组浏览器截图,显示 拟南芥 (Arabidopsis thaliana) 1 号染色体上具有多个剪接变体的基因。[@jbrowse_2016]
图:JBrowse 基因组浏览器截图,显示 拟南芥 (Arabidopsis thaliana) 1 号染色体上具有多个剪接变体的基因。[@jbrowse_2016]
数据库¶
引言¶
数据库是生物信息学的核心。在所有分析中,我们整合预先存在的数据,我们需要访问这些数据。《核酸研究》杂志每年年初出版一期关于新增和更新的数据库的专刊。这些数据库的列表也可以在线访问。
计算机科学家开发了不同类型的数据库。一个例子是关系型数据库,可以通过 SQL(结构化查询语言)进行查询,对于计算处理的数据表现良好。另一个例子是 XML(可扩展标记语言)数据库,它以指定的格式良好的 XML 文件存储数据。然而,大多数用于生物序列数据的数据库使用**平面文件数据库**,其中数据保存在结构化文本文件中。这些数据可以在文本编辑器中操作,而无需额外的数据库管理程序,并且可以在科学家之间轻松交换。缺点是搜索性能较低。这就是为什么它们通常被索引,即它们包含关键字的**索引**,类似于书中的词汇表。
根据包含的数据种类,我们区分不同类型的生物学数据库:
- **初级数据库**包含从实验获得的直接由生成数据的科学家提交的初级序列信息。
- **二级数据库**提供对初级数据库中信息的分析结果。
数据库中的每个条目都有一个唯一的**登录号**。这个号码是永久的,提供了一种明确链接到该条目的方式。登录号所指的信息不应更改。为了仍然允许更新条目,登录号可以包含一个**版本**,通常在点号之后。例如,NC_003070.9 是 拟南芥 1 号染色体在 RefSeq 中的最新版本(版本 9)。
数据库条目通常通过**交叉链接**相互链接。
延伸阅读
开发公共生物学数据库的十条简单规则 包含关于正确维护公共数据库服务所需的额外阅读材料。
GenBank¶
GenBank 是一个流行的核苷酸序列初级数据库,设在国家生物技术信息中心 (NCBI)。GenBank 版本通常每两个月发布一次,最近从 2023 年 12 月 15日发布的版本包含约 2.5 亿条序列,此外还有约 37 亿条 WGS(全基因组鸟枪法)记录。后者是尚未完成的基因组组装或基因组。完整的数据库可以通过 FTP 下载,但访问单个条目最方便的方式是通过 GenBank 网站上的搜索。
 图:GenBank 网站截图。[@genbank_2012]
图:GenBank 网站截图。[@genbank_2012]
附加信息
如今,在大多数同行评审期刊上发表文章时,科学家需要将他们的序列数据连同足够详尽的元数据(描述数据是如何产生的)一起提交给 GenBank 或相关数据库。
由于数据直接提交到 GenBank,某些基因座的信息可能高度冗余。序列记录由原始提交者所有,不能被其他人更改。
笔记 1.7:数据库冗余
在数据库上下文中,"冗余"指的是存在多次的相同数据。通常,在确定冗余时不考虑元数据。示例:两个不同的实验室确定了一个与某种疾病相关的细菌基因的 DNA 序列。元数据会不同,但序列数据是相同的,因此这两个数据库记录是冗余的。
NCBI 托管了几个被归类为"非冗余"的数据库,例如 RefSeq 非冗余蛋白质。在这里,冗余的定义是"一个非冗余蛋白质记录始终代表一个在不同菌株或物种中被观察到一次或多次的确切序列"。
GenBank 是 INSDC(国际核苷酸序列数据库合作组织)的一部分。另外两个成员数据库是 ENA(欧洲核苷酸档案库)和 DDBJ(日本 DNA 数据库)。提交到任一数据库的数据每天交换,因此所有数据库本质上包含相同的信息。
RefSeq¶
参考序列 (RefSeq) 数据集也托管在 NCBI,包含基因组 DNA、转录本和蛋白质。RefSeq 的目标是提供非冗余的、经过审核的数据。RefSeq 基因组是 GenBank 中选定组装基因组的副本。此外,转录本和蛋白质记录通过以下几种过程生成:
- 通过真核生物或原核生物注释流水线的计算。
- 人工审核。
- 从 GenBank 中已注释的基因组转移信息。与 GenBank 不同,RefSeq 记录由 NCBI 拥有,可以更新以维护注释。当前版本是 2025 年 7 月 11日发布的 231 版,包含来自约 167,000 种生物体的约 4.18 亿个蛋白质。
RefSeq 登录号直接提供关于分子类型的信息。例如,NC_ 登录号表示完整基因组,NP_ 登录号表示一个基因组中的蛋白质,WP_ 登录号表示多个基因组中的蛋白质。
UniProt¶
蛋白质有大量可用信息,例如序列信息、结构域、表达或三维结构。通用蛋白质资源 (UniProt) 的目标是提供关于蛋白质及其注释的全面资源。UniProt 包含三个数据库:
- UniProt 知识库 (UniProtKB) —— 见下文。
- UniProt 参考聚类 (UniRef) —— 在 100%、90% 和 50% 一致性水平上的蛋白质序列聚类。
- UniProt 档案库 (UniParc) —— 跨不同数据库可公开获得的蛋白质序列的非冗余档案。
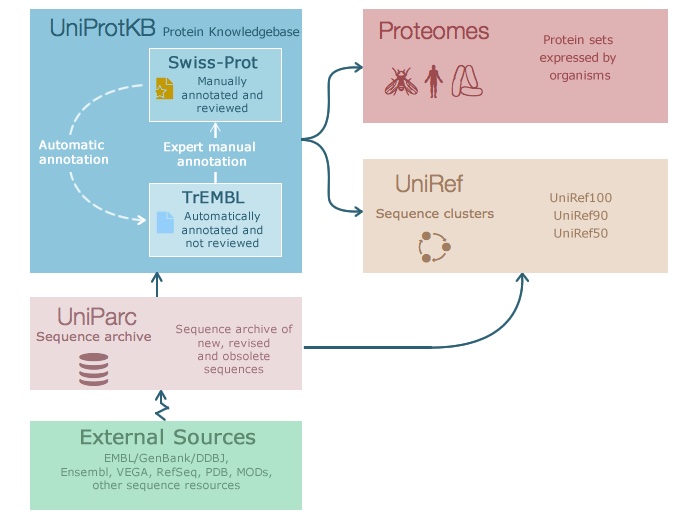 图:UniProt 中的信息流。 许可:CC BY-NC-ND 4.0 [@uniprot_2021]
图:UniProt 中的信息流。 许可:CC BY-NC-ND 4.0 [@uniprot_2021]
UniProtKB 是蛋白质功能信息的中心枢纽。对于每种蛋白质,它包含核心数据(如序列、名称、描述、分类学、引用)以及尽可能多的注释信息。它包含许多到其他数据库的交叉引用,通常是查找蛋白质信息的非常好的起点。
UniProtKB 由两个部分组成:
- Swiss-Prot —— 手工注释的记录,信息从文献中提取并经过审核的计算分析。
- TrEMBL —— 自动注释的、未经审核的记录。
UniProtKB 每 8 周更新一次。当前版本 Swiss-Prot 中约有 570,000 条条目,TrEMBL 中约有 2.5 亿条条目。
Prosite¶
Prosite 是蛋白质结构域、家族和功能位点的二级数据库。蛋白质家族中的某些区域比其他区域更保守,因为它们对蛋白质的结构或功能很重要。Prosite 包含许多蛋白质家族或结构域特有的基序和谱。在新蛋白质中搜索基序可以提供蛋白质功能的初步线索。
Prosite 的当前版本(2025 年 6 月 18日发布)包含 1311 个模式、1403 个谱和 1421 个 ProRule 条目。
Prosite 模式**通常长度为 10 到 20 个氨基酸。这些短模式通常位于高度保守的区域,如酶中的催化位点或结合位点。模式表示为正则表达式,其中氨基酸用连字符分隔,x 表示任何字母。重复也可以用括号中的重复次数给出。例如,[AC]-x-V-x(4)-{ED} 匹配包含以下氨基酸序列的序列:(丙氨酸或半胱氨酸)-任意-缬氨酸-任意-任意-任意-任意-(除谷氨酸或天冬氨酸外的任意氨基酸)。请注意,这种表示是**定性的,一个序列要么匹配一个模式,要么不匹配。
模式不能处理错配,仅限于与模式的精确匹配。因此,它们不适合识别远缘同源物。Prosite **谱**比模式更通用,也可以检测保守性较差的结构域或家族。它们在结构域的整个长度上表征蛋白质结构域,而不仅仅是保守的部分。谱从多序列比对中估计,我们将在第 2 章中了解更多。目前,重要的是知道谱模型匹配、插入和缺失。重要的是,谱是**定量**的表示,它们将返回一个序列与谱拟合程度的分数。可以应用阈值来获得序列的高分谱。与模式不同,如果序列的其余部分与谱高度相似,则可以接受与谱的错配。谱非常适合模拟结构域的结构特性。
值得注意的是,谱覆盖了结构域的结构关系,但它们也可能对一个缺乏重要功能残基的序列打高分。为了包含该信息,ProRule 包含了关于 Prosite 谱的附加信息,例如结构或功能上重要氨基酸的位置。ProRule 用于指导 UniProtKB/Swiss-Prot 的审核注释。
InterPro¶
蛋白质家族、结构域和位点综合资源 (InterPro) 将 13 个成员数据库(包括 Prosite 和 Pfam)整合为一个全面的二级数据库。此外,它还提供来自其他工具的注释,例如注释信号肽和跨膜区域。它允许通过同时使用成员数据库注释序列来识别序列中功能重要的结构域和保守位点。InterPro 可以用来查找序列属于哪个蛋白质家族,或者它的推定功能是什么。此外,一个 InterPro 条目可以整合来自成员数据库的条目(如果它们代表相同的生物实体),从而减少冗余。InterPro 条目也链接到基因本体论。它们在发布前经过审核。
InterPro 每 8 周更新一次。当前版本(2025 年 6 月 19日发布)包含约 49,000 条条目,代表不同类型。
作为一个例子,看一下 2 型苹果酸脱氢酶蛋白家族的 InterPro 条目。该条目有一个名称(苹果酸脱氢酶,2 型)和登录号 (IPR010945)。右侧显示了成员数据库中的贡献条目,带有到各个成员数据库条目的链接。描述性摘要解释了这些蛋白质是什么以及它们的功能是什么。还提供了一组 GO 术语,描述了该条目匹配的蛋白质的特征。
你可以通过运行新的序列搜索,或通过查找其 UniProt 登录号来获取蛋白质的 InterPro 注释。
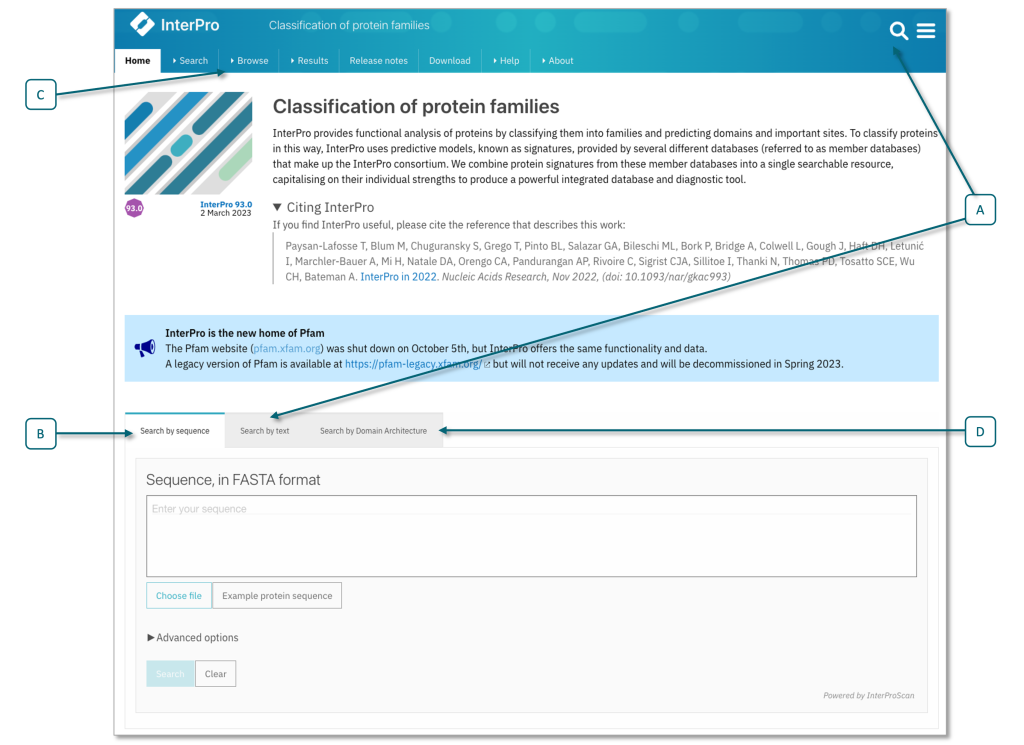 图:InterPro 主页上的搜索字段,显示文本搜索字段 (A) 和序列搜索 (B) 选项,包括"高级选项",你可以在其中将搜索限制为成员数据库或感兴趣的序列特征。在顶部菜单中选择浏览选项卡 (C) 可以访问浏览搜索(例如,搜索成员数据库签名、InterPro 条目类型),也可参见浏览页面。你还可以搜索特定的结构域架构 (D)。[@interpro_2022]
图:InterPro 主页上的搜索字段,显示文本搜索字段 (A) 和序列搜索 (B) 选项,包括"高级选项",你可以在其中将搜索限制为成员数据库或感兴趣的序列特征。在顶部菜单中选择浏览选项卡 (C) 可以访问浏览搜索(例如,搜索成员数据库签名、InterPro 条目类型),也可参见浏览页面。你还可以搜索特定的结构域架构 (D)。[@interpro_2022]
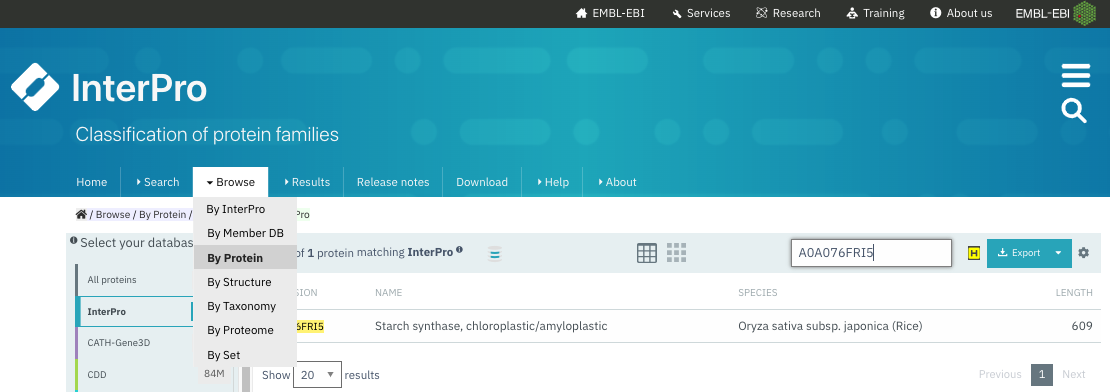 图:浏览 InterPro 中已注释的蛋白质并搜索 UniProt 登录号。[@interpro_2022]
图:浏览 InterPro 中已注释的蛋白质并搜索 UniProt 登录号。[@interpro_2022]
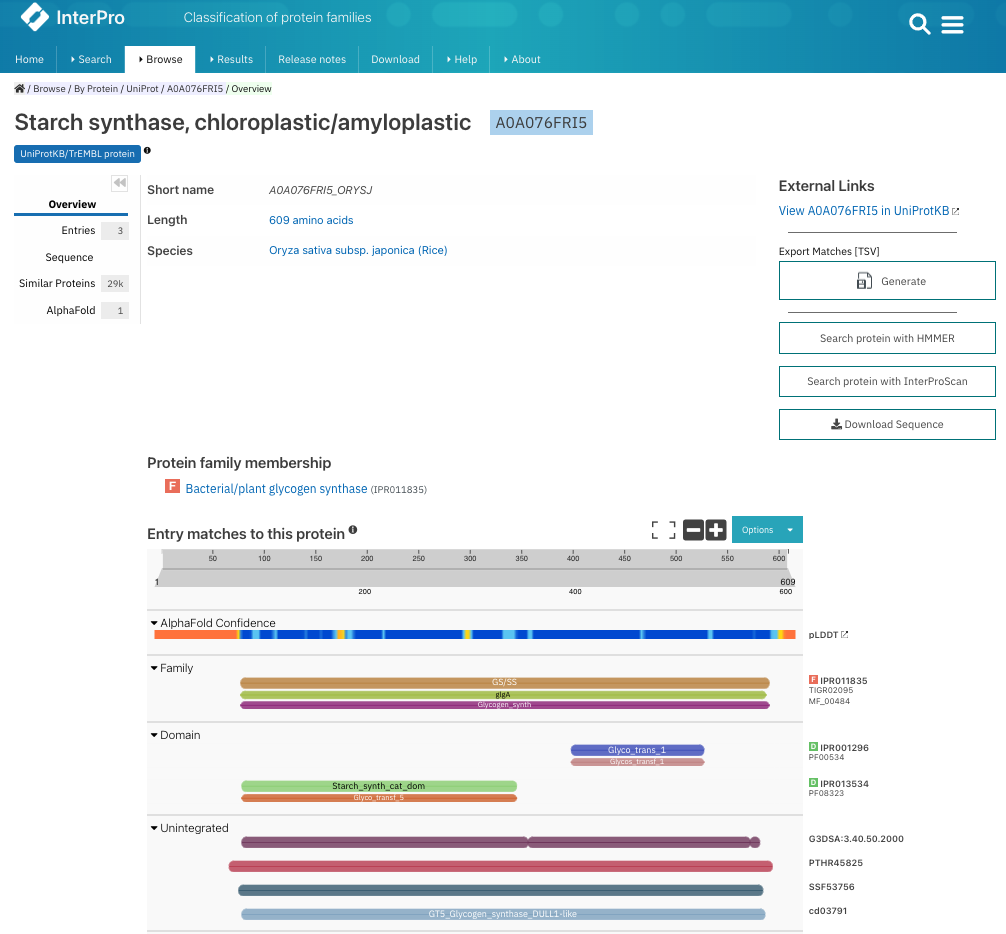 图:在 InterPro 中查找 UniProt 登录号 A0A076FRI5 的结果页面。你可以看到家族和结构域注释,右侧是 InterPro 和成员数据库中的登录号。你可以点击每个登录号来获取条目信息。[@interpro_2022]
图:在 InterPro 中查找 UniProt 登录号 A0A076FRI5 的结果页面。你可以看到家族和结构域注释,右侧是 InterPro 和成员数据库中的登录号。你可以点击每个登录号来获取条目信息。[@interpro_2022]
你可能已经注意到每个 InterPro 登录号前有一个彩色字母,例如 IPR011835 前面的 F 或 IPR001296 前面的 D。这些图标表示不同的 InterPro 条目类型:
- (同源)超家族 —— 一个大型多样化的家族,通常具有共同的蛋白质结构。
- 家族 —— 一组共享共同进化起源的蛋白质,反映在其相关功能和序列或结构相似性上。
- 结构域 —— 蛋白质中独特的功能性或结构单元,通常负责特定功能或相互作用。
- 重复 —— 通常是在蛋白质中重复的短氨基酸序列。
- 位点 —— 具有某些特征的一组氨基酸,可能对蛋白质功能重要,例如活性位点或结合位点。
 图:不同 InterPro 条目的图标(同源超家族、家族、结构域、重复或位点)。 许可:CC BY-SA 4.0 [@interpro-types_2020]
图:不同 InterPro 条目的图标(同源超家族、家族、结构域、重复或位点)。 许可:CC BY-SA 4.0 [@interpro-types_2020]
延伸阅读
你可以在这里找到有关 InterPro 条目类型及示例的更多信息。
Pfam¶
Pfam 是蛋白质结构域的重要资源。在 Pfam 中,结构域根据建模为隐马尔可夫模型 (HMM) 的谱进行分类。我们将在第 2 章中学习更多关于 HMM 的知识。Pfam 现已集成到 InterPro 中。每个 Pfam 结构域可以用一个 logo 表示,其中在特定位置出现频率较高的氨基酸用较大的字母表示。
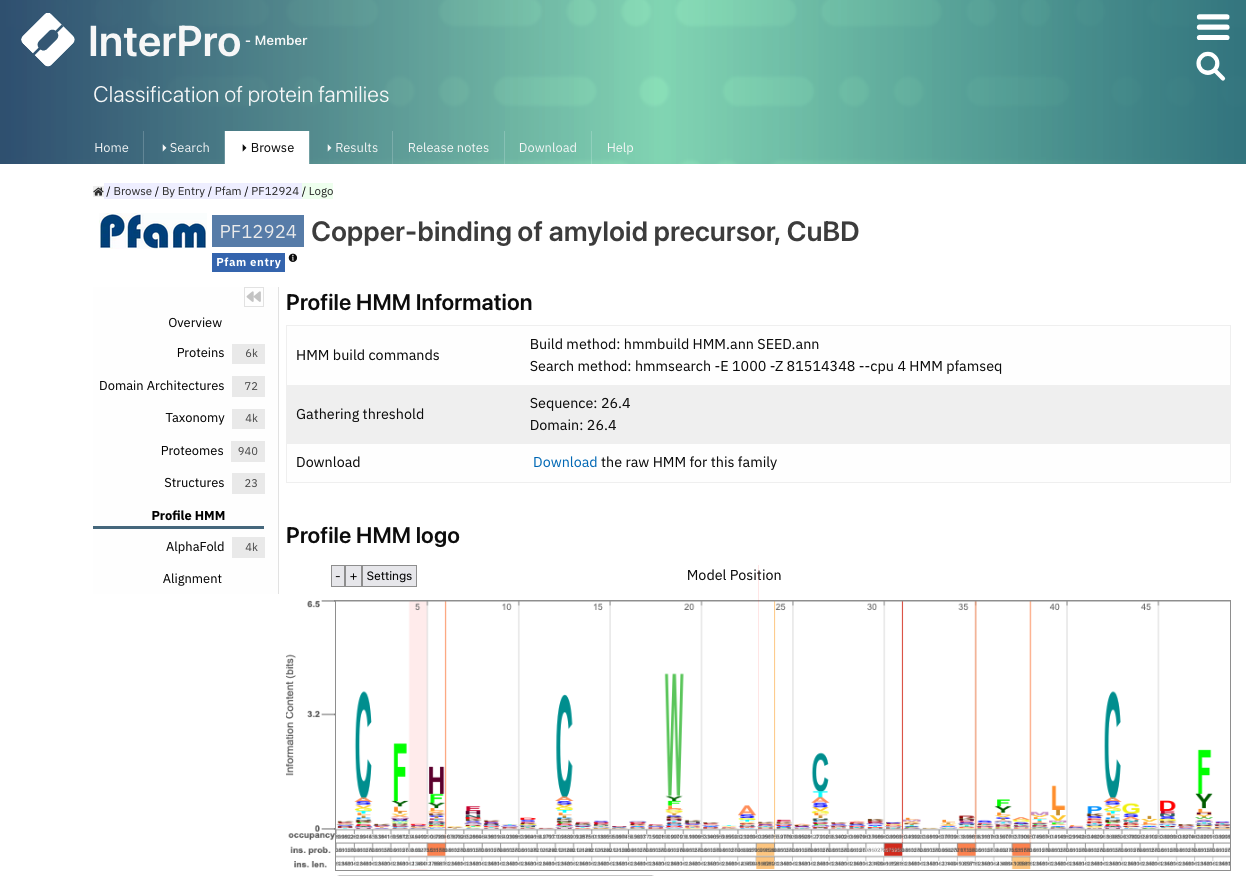 图:PF12924 的 Pfam logo。[@interpro_2022]
图:PF12924 的 Pfam logo。[@interpro_2022]
文件格式¶
生物数据有许多不同的格式。格式是关于数据内容和组织的一组规则。你应该熟悉生物信息学中的几种常见数据格式,这些将在实践课中使用。
| 文件格式 | 用途 | 常见扩展名 |
|---|---|---|
| FASTA | 核苷酸或氨基酸序列 | .fa, .fasta, .fna, .faa |
| GenBank | 序列、注释、元数据 | .gb |
| 通用特征格式 | 序列注释 | .gff |
| FASTQ | DNA 测序数据,包括碱基识别质量分数 | .fq |
| SAM/BAM(序列/二进制比对和映射) | 通常是原始测序数据与参考序列之间的比对,附带质量分数 | .sam(.bam 为二进制文件格式) |
| VCF(变异调用格式) | 基于与参考序列比对的测序数据的(遗传)变异调用 | .vcf(.bcf 为二进制文件格式) |
| PDB | 蛋白质结构数据 | .pdb |
| 非结构化文本 | 杂项 | .txt |
纯文本文件¶
许多生物数据格式是纯文本文件:它们只包含字母、数字和符号,但没有格式(如字体大小或颜色)。虽然这些文件类型可以有各种不同的扩展名(例如 .fasta、.gff 等),但它们只包含纯文本。纯文本文件的优点是可以用任何计算机上的任何文本编辑器打开。纯文本与富文本格式不同,后者还可以包含格式。许多生物信息学程序期望纯文本文件作为输入。因此,在计算机上创建它们时,注意保存为这种格式,而不是例如 rtf 或 word 格式。
在 Windows 计算机上,纯文本文件可以使用记事本程序创建。
 图:Windows 上记事本的截图。 许可:CC BY-NC 4.0 [@own_1_2024]
图:Windows 上记事本的截图。 许可:CC BY-NC 4.0 [@own_1_2024]
在 Mac 上,纯文本文件可以使用 TextEdit 程序创建。注意将设置设为纯文本。
 图:Mac 上 TextEdit 的截图。 许可:CC BY-NC 4.0 [@own_1_2024]
图:Mac 上 TextEdit 的截图。 许可:CC BY-NC 4.0 [@own_1_2024]
参见
如果你还不熟悉纯文本编辑器,现在就试试,在计算机上编写并保存一个纯文本文件吧!
生物信息学中有一些重要的文件格式。一个 **FASTA 文件**存储 DNA 或蛋白质序列。关于序列的信息在头部(以 > 开头),头部在一行上,序列可以跨越多行。一个多 FASTA 文件存储多条序列。
 图:FASTA 格式的序列。 许可:CC BY-NC 4.0 [@own_1_2024]
图:FASTA 格式的序列。 许可:CC BY-NC 4.0 [@own_1_2024]
**GenBank 文件格式**是表示基因或基因组的流行格式。这里你可以找到一个带有注释的 GenBank 记录示例。重要元素包括基因座 (Locus)、定义 (Definition)(即名称)和生物体 (Organism)。此外,特征 (Features) 如基因和 CDS(编码序列)也被列出。
二进制文件¶
二进制文件是所有不是文本文件的文件,不能在文本编辑器中打开。它们需要特殊的程序来编写、打开和解释。示例包括 Word 文件 (.docx),可以用 Word 打开;PDF 文件 (.pdf),可以用 Acrobat Reader 打开;或图像文件(例如 .png),可以用图像查看器打开。
二进制文件有时也在生物信息学中使用。示例包括 bam 格式,它是 sam 格式的二进制版本,或 gzip 格式。Gzip 用于在不丢失信息的情况下压缩文本文件。对于大文件,这种方式可以节省大量磁盘空间。
本体论¶
本体论 (Ontology) 是针对特定领域(如生物学、遗传学或医学)的全面且结构化的词汇表。它定义了领域中使用的各种术语,以及它们的含义和相互关联。因此,本体论作为标准化框架,以有效沟通和推理的方式组织和分类信息,为研究人员、从业人员和计算机系统提供服务。例如,本体论中的术语可以涵盖基因、蛋白质和细胞等生物学实体,以及发生在活生物体中的过程、功能和相互作用。本章提到的大多数数据库都以某种方式使用本体论来描述其数据。
本体论在生物信息学中起着至关重要的作用,因为它们促进了:
- 标准化和一致性:本体论为研究人员和专业人士提供了共同的语言和一致的框架,确保每个人都以相同的方式理解和使用术语。
- 互操作性:本体论促进了跨不同研究组、机构和数据库的数据和知识的共享与整合。它们使计算机系统能够更准确地处理数据,从而进行更有意义的分析和发现。
- 科学推理:通过以逻辑和结构化的方式组织信息,本体论帮助研究人员更有效地生成假设、设计实验和验证发现。
笔记 1.9:FAIR 原则
如上所述,本体论促进了科学的可重复性。科学可重复性的一个关键概念是 FAIR 原则,FAIR 代表可发现 (Findable)、可访问 (Accessible)、可互操作 (Interoperable) 和可重用 (Reusable)。本教材不详细描述它们,但你应该阅读以下在线资源以熟悉 FAIR 原则。
本体论通常形成层级结构,其中特定术语指向更通用的术语。更一般地说,大多数本体论表示为图,其中本体论术语是节点,术语之间的关系是边。因此,一个本体论术语可以有多个父术语。生命科学中经常使用多种本体论,下面将详细讨论其中一些。
基因本体论¶
基因本体论 (GO) 是基因和基因产物(例如蛋白质)功能的知识库。它组织为三个不同的领域,涵盖各个方面:
- 分子功能:基因产物(例如蛋白质)执行的分子层面功能,如"催化"或"转运"。大多数分子功能可以由单个基因产物执行,但有些功能由多个(可能不同的)基因产物组成的复合体执行。GO 分子功能通常包含"活性"一词(淀粉酶_酶将具有 GO 分子功能_淀粉酶活性)。
- 细胞组分:基因产物执行其功能的细胞结构(或相对于它们的定位)。可以是细胞区室(例如线粒体)或它们所属的大分子复合体(例如核糖体)。
- 生物过程:由多个分子活动组成的较大的生物程序,例如 DNA 修复 或 信号转导。
笔记 1.10:分子通路?
生物过程不等同于分子通路。目前,基因本体论不代表完全描述通路所需的动态或依赖关系。
一个展示本体论如何表示为图的好例子是生物过程己糖生物合成过程,它有两个父节点:己糖代谢过程和单糖生物合成过程。这反映了生物合成过程是代谢过程的子类型,己糖是单糖的子类型。
GO 层级中 GO 术语之间的边可以表示基因和基因产物之间的各种关系。基因本体论中使用的四种主要关系类型是"是一种"、"部分"、"具有部分"和"调控"。
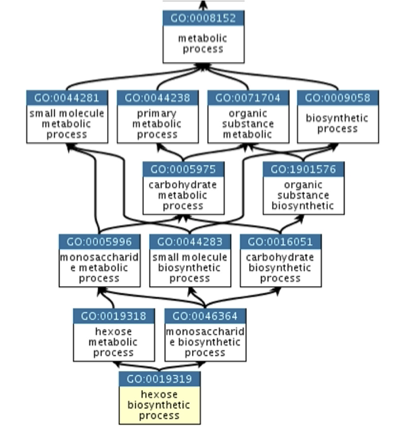 图:基因本体论层级的摘录。[@go_2009]
图:基因本体论层级的摘录。[@go_2009]
序列本体论¶
序列本体论 (SO) 描述了基因或重复序列等生物序列元件,以及它们的特征和属性。
序列本体论组织在四个主要层级:
- 属性 (Attribute):属性描述给定序列的某种性质,例如序列来源(即如何产生的)。
- 集合 (Collection):多个不连续的序列在一起,例如完整基因组的染色体。
- 特征 (Feature):最通用的顶级条目,描述连续生物序列的任何范围,例如基因是一个区域,而区域反过来又是一个序列特征。
- 变体 (Variant):旨在描述遗传变异。序列变体的定义由序列本体论中的其他条目组成:"一个序列变体是序列特征或基因组的非精确拷贝,表现出一个或多个序列改变"。
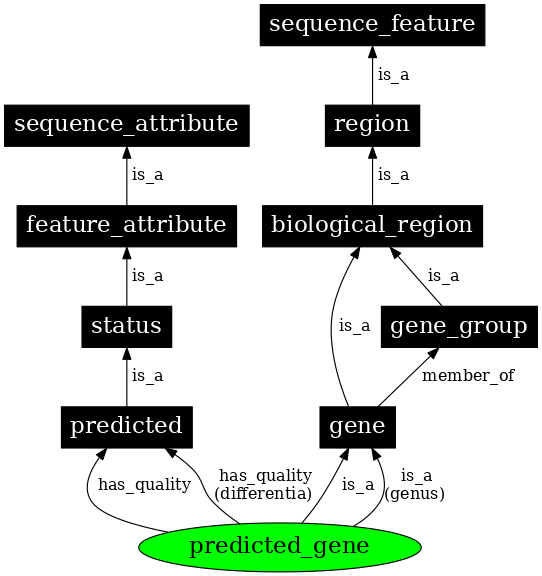 图:序列本体论层级的摘录。[@so_2005]
图:序列本体论层级的摘录。[@so_2005]
其他本体论¶
还存在许多与生物医学研究相关的本体论。欧洲生物信息学研究所 (EBI) 提供了本体论查询服务,便于搜索本体论。其他本体论的例子包括描述植物各种解剖结构的植物本体论,以及人类疾病本体论。
实践作业¶
本实践包含帮助你消化第 1 章学习材料的问题和练习。你有 2 个上午的时间来完成这些练习。在单个会话中,你应该目标完成本指南的大约一半(即第 1 天:作业 1-3,第 2 天:作业 4 和项目准备练习)。利用时间提示确保你不会卡在一个作业上。这些实践练习为你提供了项目的最佳准备。特别是末尾的**项目准备练习**很好地反映了撰写优秀项目报告所需的水平。确保你现在就培养实践技能,以便在项目期间应用它们。
注意,答案将在实践课后公布!
DNA/基因,45 分钟
-
你如何区分核糖和脱氧核糖?
-
哪些碱基是嘌呤?
-
A 的互补碱基是什么?C 呢?G 呢?T 呢?
-
序列 ACGGTGATC 的反向互补是什么?
-
序列 ATCGATCGGC 的 GC 含量是多少?
-
以下哪项正确?核苷酸序列的书写方向为:\ A. 5' 到 3' \ B. 3' 到 5'
-
在 DNA 序列中,G 代表:\ A. 甘氨酸 \ B. 鸟嘌呤 \ C. 葡萄糖 \ D. 谷氨酸
-
给定一条编码 DNA 链。写出非编码链、转录序列和产生的氨基酸链。你可以参考复制过程图。
编码链: 5' ATGGTTTTACTTGAA 3' 非编码链: ...................... mRNA: ...................... 氨基酸: ...................... -
在你的计算机上,浏览到 UniProt 并搜索 UniProt ID B3H4Y2。\ a. 这种蛋白质在哪种生物体中?这种蛋白质的长度是多少?对应的基因 ID 是什么?\ b. 写出该蛋白质的前 5 个和后 5 个氨基酸。
-
浏览到 arabidopsis.org 并点击 "JBrowse"(推荐使用 Firefox 或 Chrome)。这将带你到 拟南芥 基因组的基因组浏览器。搜索问题 9 中的基因 ID。在 "Help" -> "General" 下你可以找到一些帮助你理解你正在查看内容的信息。\ a. 你可以看到这个基因产生两种不同的 mRNA 转录本(用 .1 和 .2 标示),因此产生 2 种不同的蛋白质。这些转录本包含多少个外显子?多少个内含子?\ b. 打开 "RNA-seq based evidence"/"Aligned reads" 下的 "Light grown seedling" 轨道。你能识别剪接位点吗?内含子的前两个和后两个碱基是否符合预期?\ c. 保存转录本 1 的数据。保存整个转录本的 fasta 文件和每个编码序列 (CDS) 的 fasta 文件。在计算机上创建一个包含该蛋白质完整编码序列的 fasta 文件。\ d. 编码序列的长度是否与你的预期一致(基于你在问题 9a 中的发现)?\ e. 翻译前几个和后几个密码子,与蛋白质序列(问题 9)进行比较。它们匹配吗?\ f. 查看基因上游。你能找到 TATA box 吗?在转录开始之前有多少个核苷酸?
-
GC 含量\ a. 在网上找一个工具来计算基因的 GC 含量。你找到了哪个工具?用它来计算你在上一任务中创建的整个转录本和编码序列的 GC 含量。你观察到了什么?\ b. 查找该基因所在染色体的 GC 含量(提示:在 NCBI Genome 中搜索该物种)。阅读编码序列中的 GC 含量。这里呈现的哪些信息与你的分析一致?
-
为什么病毒不在生命之树中表示?看看这个网站。
-
浏览到 NCBI 分类学数据库。查找以下物种的域和科:
| 物种 | 域 | 科 |
|---|---|---|
| Moraxella catarrhalis | ||
| Haloarcula quadrata | ||
| Loxodonta cyclotis |
蛋白质,45 分钟
- 氨基酸甘氨酸有什么特别之处?
- 列出三种疏水性氨基酸。
-
哪些氨基酸是酸性的? | | | | | |--|--|--|--| | alanine | glutamine | leucine | serine | | arginine | glutamic acid | lysine | threonine | | asparagine | glycine | methionine | tryptophan | | aspartic acid | histidine | phenylalanine | tyrosine | | cysteine | isoleucine | proline | valine |
-
以下哪项不正确?\ a. A = 精氨酸 (Arginine)\ b. V = 缬氨酸 (Valine)\ c. Q = 谷氨酰胺 (Glutamine)\ d. T = 苏氨酸 (Threonine)
-
在折叠的蛋白质中,非极性氨基酸倾向于:\ a. 在蛋白质内部\ b. 在蛋白质表面\ c. 随机分布
-
氨基酸的侧链在蛋白质的折叠和功能中起着重要作用。下面你可以看到一个由五个氨基酸形成的短肽(标记为 1 到 5)。\ a. 分别用蓝色和红色标出肽的 N 端和 C 端,并用绿色高亮所有肽键。\ b. 对于五个氨基酸(1-5)中的每一个,根据缺少的信息给出名称、三字母或一字母缩写(例如,对于氨基酸 1,给出三字母和一字母缩写,对于氨基酸 2 给出名称和三字母缩写)。\ c. 指出每个氨基酸(1 到 5)的物理化学性质(非极性、极性、酸性、碱性)。\ d. 用一句话描述氨基酸 4 侧链的特定性质,以及为什么这个性质对形成蛋白质结构很重要。
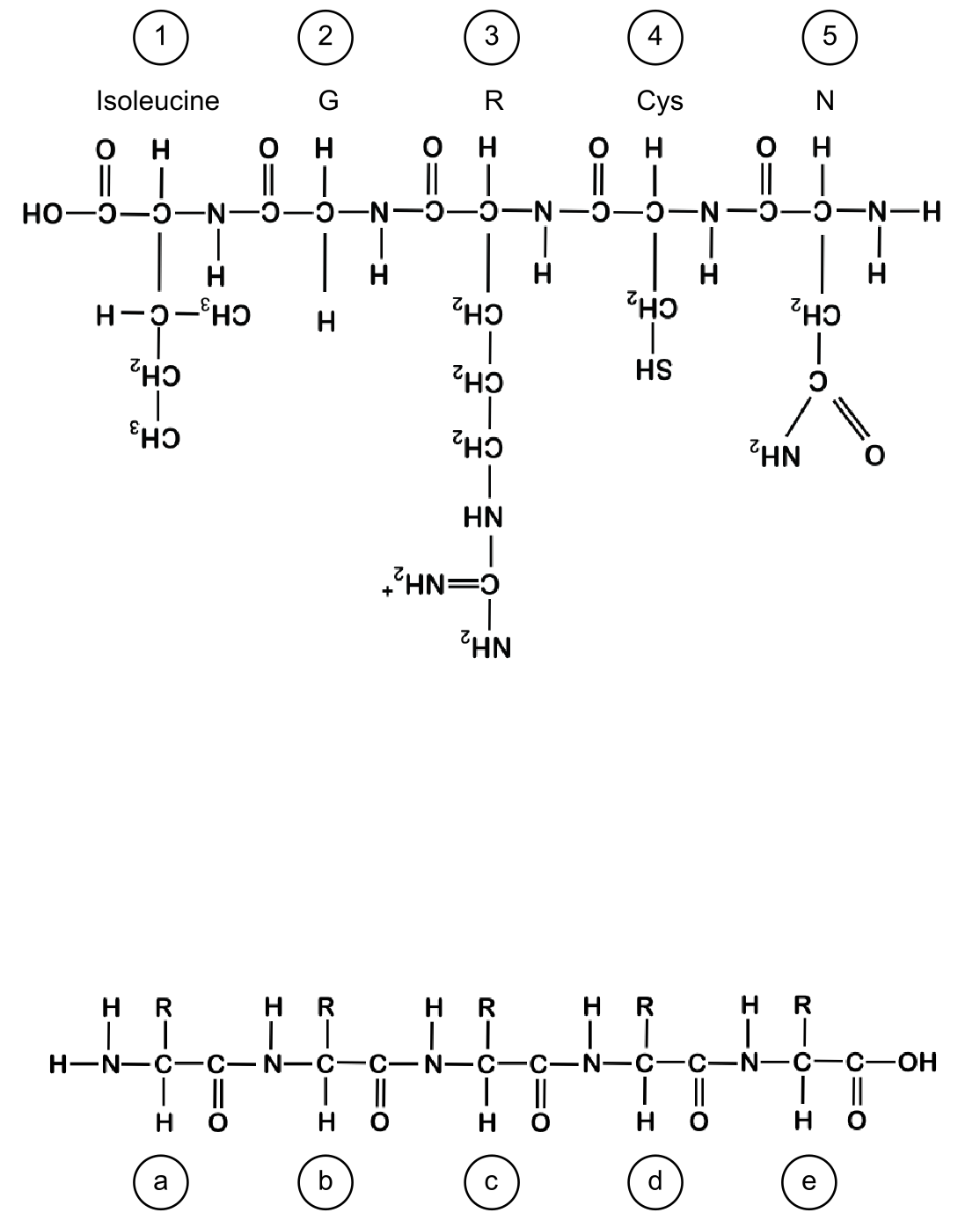 图:由五个氨基酸组成的短肽。
图:由五个氨基酸组成的短肽。
-
氨基酸及其侧链可以与其他氨基酸相互作用并形成键和相互作用。氨基酸及其侧链之间的相互作用在稳定的折叠蛋白质结构中起着重要作用。回顾三级结构相互作用图。根据这些信息,再看一下具有五个氨基酸的肽序列(见问题 6)。在此肽下方,你将看到另一个肽的骨架,其侧链仅用 R 表示(标记为 a-e)。查看教材中的 20 种氨基酸。与你的同伴讨论,20 种可能的氨基酸中哪些可以放置为侧链 R (a-e),使得它们可能与上方肽中相应的氨基酸 (1-5) 相互作用(即形成键或其他相互作用),即 a 与 1 相互作用,b 与 2 相互作用,以此类推。指出你提出的每对氨基酸之间发生了哪种类型的相互作用(例如氢键)。
-
蛋白质折叠为紧凑的结构,这种结构对于蛋白质具有生物功能活性很重要。在折叠的蛋白质(三级结构)中,二级结构通常仍然可见,即螺旋和 β-折叠仍然可见。有时蛋白质不仅由单个结构单元(所谓的结构域)组成,而是由多个可以是相同类型或不同类型的结构域组成。有时,查看具有已知折叠的蛋白质的三级结构(通过实验或计算方法确定)可能很有用,例如查看结构中的突变发生在哪里。我们将查看 Gamma B-晶状体蛋白的蛋白质结构。前往 PDB 网站,这是一个蛋白质结构资源,在主页上搜索 Gamma B-晶状体蛋白,ID 为 "1AMM"。点击 '3D View' 查看结构的三维模型。在右下角,将查看器更改为 NGL。 a. 按二级结构着色(见 'Color',在 'Structure View' 下)。在 'Structure View Documentation' 下你可以找到每种颜色的含义。你能识别结构中可以观察到的二级结构元件(螺旋、折叠)的数量吗? b. 这个蛋白质有多少个结构域?
-
氨基酸测验:你现在已经广泛使用了氨基酸,你应该了解 1 字母和 3 字母缩写之间的关系、氨基酸的名称及其生化性质。为了再次测试这些知识,填写表中缺少的信息(在完成测验之前不要看教材)。
| # | 1 字母 | 3 字母 | 全名 | 类别 |
|---|---|---|---|---|
| 1 | 谷氨酸 (Glutamic acid) | 非极性/极性/酸性/碱性 | ||
| 2 | Phe | 非极性/极性/酸性/碱性 | ||
| 3 | T | 非极性/极性/酸性/碱性 | ||
| 4 | Pro | 非极性/极性/酸性/碱性 | ||
| 5 | 丝氨酸 (Serine) | 非极性/极性/酸性/碱性 | ||
| 6 | K | 非极性/极性/酸性/碱性 | ||
| 7 | 异亮氨酸 (Isoleucine) | 非极性/极性/酸性/碱性 | ||
| 8 | Asn | 非极性/极性/酸性/碱性 | ||
| 9 | 甲硫氨酸 (Methionine) | 非极性/极性/酸性/碱性 | ||
| 10 | A | 非极性/极性/酸性/碱性 | ||
| 11 | P | 非极性/极性/酸性/碱性 | ||
| 12 | His | 非极性/极性/酸性/碱性 |
数据库,45 分钟
-
在网页浏览器中,导航到 核酸研究 杂志的分子生物学数据库合集 (NAR)。从列表中选择三个引起你注意的数据库,最好来自不同类别,并探索它们(每个约 5 分钟)。\ a. 里面有什么类型的数据?\ b. 它用于什么?高度专业化还是广泛的应用?\ c. 如何搜索该数据库?\ d. 它看起来是最新的且定期维护的吗?
-
冗余\ a. 数据库中的冗余是什么意思?给出序列数据库中冗余的例子。\ b. UniProt 数据库是冗余的还是非冗余的?\ c. RefSeq 和 GenBank 在冗余方面有什么区别?
-
本体论\ a. 描述什么是本体论(使用教材中的信息和/或 Google 查找信息)。\ b. 基因本体论是生物信息学中最重要的本体论之一。基因本体论涵盖了哪些生物学领域?\ c. 在 UniProt 中查找 拟南芥 蛋白质(登录号 B3H4Y2)。你找到了关于与该蛋白质相关的 GO 术语的什么信息?\ d. 现在查找著名的 拟南芥 基因 FRIGIDA(登录号 P0DH90)。哪些 GO 术语与该基因相关?该蛋白质在哪个细胞组分中被发现,参与哪个生物过程?
-
UniProt\ a. 在 UniProt 中再次查找问题 3 中的两种蛋白质。它们分别存放在 UniProt 的哪个部分?两者中哪个具有更高的注释质量?\ b. 每种蛋白质链接了多少篇文献?这些文献中哪些包含关于蛋白质的具体信息(根据标题判断)?\ c. 对于每种蛋白质,查找至少一个你已知的数据库的交叉引用和一个你还不知道的数据库的交叉引用。花几分钟浏览通过这种方式获得的信息。\ d. 使用 PIR 网站计算两种蛋白质序列中各氨基酸的频率。你注意到什么值得注意的地方了吗(提示:查看各种氨基酸的相对丰度)?你能将此与 UniProt 中存在的信息联系起来吗(提示:查看家族/结构域)?
-
生物数据管理中的一个热门话题是"FAIR"数据。FAIR 中的字母代表什么,这些术语是什么意思?
基因组注释,120 分钟
- 解释同源性搜索在基因组注释中如何有用,以及为什么它对真核生物比对原核生物更复杂。
- RNA 测序数据如何帮助基因预测?仅靠 RNA 测序数据本身是否足以注释基因组?
-
在 UniProt 数据库 中查找标识符为 B3H4Y2 的 拟南芥 蛋白质。对应的基因 ID 是 AT1G65484。在第二个标签页中,在 TAIR JBrowse 中查找该基因。该基因的序列可以在 BrightSpace 上找到。在第三个标签页中,前往 NCBI 开放阅读框查找器 并以 FASTA 格式粘贴基因序列并点击提交。\ a. 找到了多少个 ORF?它们都在同一阅读框中吗?\ b. 是否有任何 ORF 对应于已注释基因中的 ORF?为什么是或为什么不是?\ c. 简单的 ORF 检测工具什么时候有用?什么时候不够用?
-
酵母 (酿酒酵母,Saccharomyces cerevisiae) 是一个被广泛研究的模式生物。大量关于酵母的信息存储在 酿酒酵母基因组数据库 (SGD) 中。你将使用参考菌株 S288C。酵母相对较小的基因组使我们有在线探索一些基因组注释工具(在合理时间内)并将我们的发现与可用的高质量注释进行比较的机会。在 SGD 菜单中点击 Sequence -> Reference Genome -> Genome Snapshot。在 "Features by Type" 下探索图形和表格视图。\ a. 酵母基因组包含多少个基因?\ b. 3 号染色体上已注释了多少个 tRNA?线粒体基因组上有多少个?\ 首先你将使用 MITOS2 注释酵母的线粒体基因组。\ c. 使用 Brightspace 上提供的线粒体基因组或从 SGD 下载序列。线粒体 (MT) 基因组的长度是多少?发挥创意或使用 Google 来找到答案。MT 基因组是线性的还是环形的?\ d. 酵母 MT 基因组不使用标准遗传密码。它使用的是哪一个?使用 NCBI 遗传密码 来找到答案。有多少个密码子具有不同的含义?\ e. 前往 MITOS2 工具。选择与酵母相关的参考序列和遗传密码并上传 fasta 文件。确保将输出更改为 GFF。点击提交。此注释大约需要 10 分钟,因此继续下一个练习(5 和 6)直到结果完成。\ f. 通过点击浏览结果。预测了多少个 tRNA?\ g. 在 SGD 上的基因组浏览器中查找第一个预测的 tRNA。预测是否与已知 tRNA 匹配?该 tRNA 对应哪种氨基酸?与此 tRNA 基因相关联的 GO 术语有多少个?\ h. 关于 "giy" 基因你能发现什么?在 FAA fasta 文本文件(从左侧菜单下载)中你可以找到该基因产生的蛋白质序列。前往 InterPro 并使用蛋白质序列通过 InterProScan 搜索已知的蛋白质结构域/功能。你找到了该蛋白质的什么功能信息?(运行 InterProScan 可能需要几分钟,同时继续下一个问题)。
-
接下来,我们将使用广泛使用的 Augustus 从头基因预测器 在 3 号染色体上预测蛋白编码基因。Augustus 以 GFF3 文本格式输出其预测。为了理解 Augustus 的输出,请先在这里熟悉该格式。Augustus 基因预测器已经在许多生物上训练过。\ a. 上传 3 号染色体的序列,选择正确的生物并"运行 AUGUSTUS"(预测两条链上的基因)。预测了多少个基因?其中多少个包含内含子?\ b. 关于基因 g100 的功能、结构域等你能说出什么?你使用了哪些数据库来查找这些信息?
-
你现在已经看到了基因组注释中使用的几个工具示例。用自己的话描述结构基因组注释和功能基因组注释,并各给出一个例子。
项目准备练习
我们希望获得关于 拟南芥 中 ARF 基因家族成员的见解。ARF5(UniProt ID P93024)和 IAA5(UniProt ID P33078)是两个被广泛研究的 拟南芥 蛋白质,在生长素介导的基因表达调控中发挥作用。因此,它们被选为探索植物 ARF 基因家族的起点。 对 ARF5 和 IAA5 进行小型背景研究。 探索 ARF5 和 IAA5 的蛋白质序列、性质(如长度、组成等)、相互作用伙伴和功能区域。 最后,探索 拟南芥 中编码 ARF5 和 IAA5 的基因(基因组位置、外显子结构、表达等)。 用几个要点分别描述以下项目。你可以包含最多两张图表。
- 材料与方法 你做了什么?你使用了哪些数据、数据库和工具,为什么选择这些?你选择了哪些重要设置?
- 结果 你发现了什么,主要结果是什么?报告相关数据、数字、表格/图表,并清楚描述你的观察结果。
- 讨论与结论 结果合理吗?它们是否符合同你的预期,还是你看到了令人惊讶的地方?结果意味着什么,你如何解释它们?不同工具是否一致?你能得出什么结论?确保描述你解释背后的预期和假设。
参考文献¶
{{ bibliography }}
术语表¶
术语表
- 注释 (Annotation)
- 识别基因组序列中功能元件(如基因、编码区域和调控基序)的过程。
- 细胞 (Cell)
- 所有生物体的基本结构和功能单元。
- DNA
- 脱氧核糖核酸 (**D**eoxyribo**N**ucleic **A**cid)
- 外显子 (Exon)
- 基因中的一个 DNA 片段,在内含子去除后编码成熟信使 RNA (mRNA) 的一部分。
- 基因 (Gene)
- 编码功能产物(通常是蛋白质)的 DNA 片段。
- 基因组 (Genome)
- 存在于细胞或生物体中的全部基因或遗传物质。
- 基因组浏览器 (Genome browser)
- 用于可视化检查基因组区域、注释和实验数据轨道的工具。
- HMM
- 隐马尔可夫模型 (Hidden Markov Model) —— 一种统计模型,表示状态不可直接观察(隐藏)但可以从观测数据推断的系统。
- 内含子 (Intron)
- 基因中的一个 DNA 片段,不在成熟信使 RNA (mRNA) 产物中表达,在 RNA 剪接过程中被去除。
- mRNA
- 成熟 mRNA (mature messenger RNA),是一种经过处理的 RNA 形式,已去除内含子,仅由外显子组成,准备好翻译为蛋白质。
- miRNA
- 微小 RNA (MicroRNA),是含有 21-23 个核苷酸的小型、单链、非编码 RNA 分子。
- 核苷酸 (Nucleotide)
- DNA 和 RNA 的基本构建块,由碱基、糖和磷酸基团组成。
- 蛋白质 (Protein)
- 由氨基酸组成的分子,由基因编码,负责细胞的结构和功能。
- RNA
- 核糖核酸 (RiboNucleic Acid)
- rRNA
- 核糖体 RNA (ribosomal RNA),是一种非编码 RNA,是所有活细胞中蛋白质合成所必需的核糖体的关键组成部分。
- 序列 (Sequence)
- DNA 或 RNA 链中核苷酸的精确排列顺序。
- 剪接 (Splicing)
- 一种生物过程,从前体信使 RNA (pre-mRNA) 转录本中去除非编码区域(内含子),并将编码区域(外显子)连接在一起,形成可以翻译为蛋白质的成熟信使 RNA (mRNA)。
- 转录 (Transcription)
- 将 DNA 序列复制为 RNA 的过程。
- 翻译 (Translation)
- 将 RNA 序列转换为蛋白质的过程。
- tRNA
- 转运 RNA (Transfer RNA),是一种帮助将信使 RNA (mRNA) 序列解码为蛋白质的 RNA 分子。
本站总访问量 次