第三章
课程来源与翻译声明
- 原著版权:本教程翻译自瓦赫宁根大学(Wageningen University & Research)的经典开源课程 Introduction to Bioinformatics。
- 翻译说明:本版本主要采用人工智能(机器翻译)进行全文本地化,并由团队进行了初步校对。部分专业术语可能存在翻译不够地道或准确的地方。
- 勘误反馈:若你在学习过程中遇到语病、错别字、代码失效或概念歧义,欢迎随时点击页面右上角的 :material-edit: 编辑此页(或联系课题组/在 GitHub Issue 中)提交反馈,帮助我们持续完善本教程。
在本章中,你将学习系统发育树是什么、如何解读这些树,以及如何从 DNA 或氨基酸序列的多重序列比对中推断它们。
学习目标
学习本章后,你应该能够: - 描述系统发育树的目的与结构。 - 解读多重序列比对(MSA)中的特征与特征状态。 - 从系统发育树中推断进化关系。 - 解释基因树与物种树之间的区别。 - 评估系统发育树的分辨率并识别多分支。 - 应用树的根化概念来确定进化方向。 - 比较主要的建树方法。 - 使用替换模型准确估计序列分歧度。 - 将 Bootstrap 分析解读为节点支持度的衡量指标。 - 使用 Newick 格式和树工具读取、写入和可视化系统发育树。
简介¶
在本章中,你将学习使用多重序列比对(MSA)(如你在第 2 章中构建的那些),并将其所包含的变异信息可视化为系统发育树。系统发育树被认为是一种高效的数据结构,能够概括你的 MSA 中的数据及其变异信息。系统发育树由特征(characters)构建,特征即 MSA 中的各个列或位置。特征具有状态(states),在本例中为该位置上发生的各个核苷酸或氨基酸替换(见下文的特征与系统发育树)。不变特征(invariable characters)是仅被一种核苷酸或氨基酸"占据"的列或位置,而可变特征则可能包含多达 4 种不同核苷酸或 20 种氨基酸。
DNA 和氨基酸(AA)序列包含构建蛋白质结构所需的信息。在 MSA 中比较它们可以深入了解这些结构及其相关功能如何自从祖先序列分化以来在进化过程中发生变化。序列之间的特征状态变化(即替换)越多,它们就越分歧,亲缘关系可能也越远(见下文的相关与分歧),因此在系统发育树上的距离也越远。系统发育树中的信息本质上是层级性的,即由嵌套的子树集合构建而成,这些子树也称为分支(clades)。分支(clade)是一个包含祖先及其所有后代的群体,也称为单系群(monophyletic group)。
基本原理¶
简化的生命之树。来源:CC BY 4.0 [@tree_of_life_alt_2014]
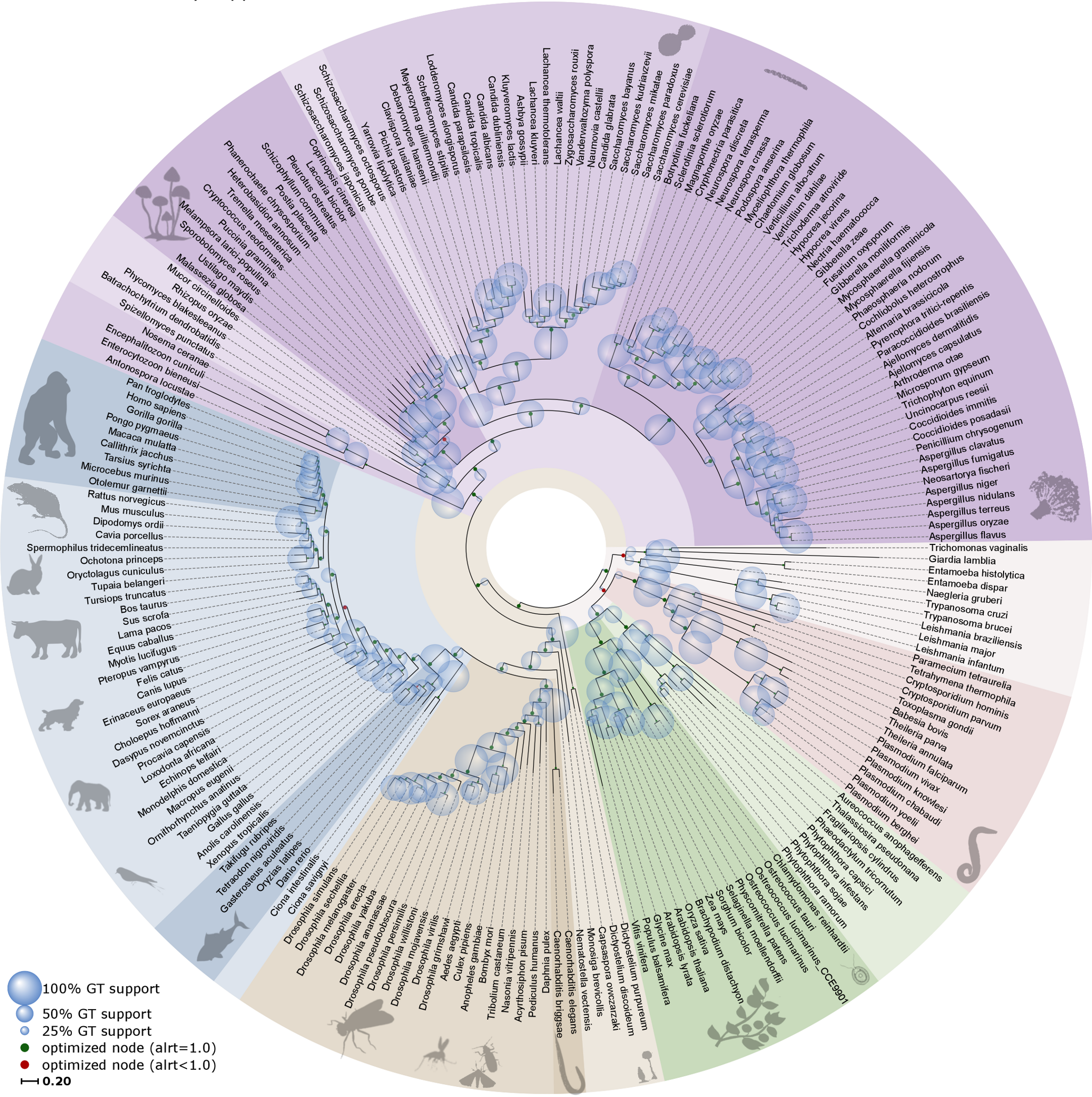
为什么我们应该研究系统发育学(Phylogenetics)?它究竟是什么?自达尔文以来,我们知道所有生物都相互关联,形成一幅生命画卷,构成一棵包含万物的系统发育树。系统发育学旨在理解基因、物种和更高分类单元之间的进化关系,因此几乎与所有生物学问题都密切相关。为什么?因为进化背景(而非"快照"视角)使我们能够识别进化谱系及其起源,并提供有关生命形式和序列如何在数百万年间变化和适应的信息。例如研究基因组内基因家族的进化,或谱系中物种关系的构建。
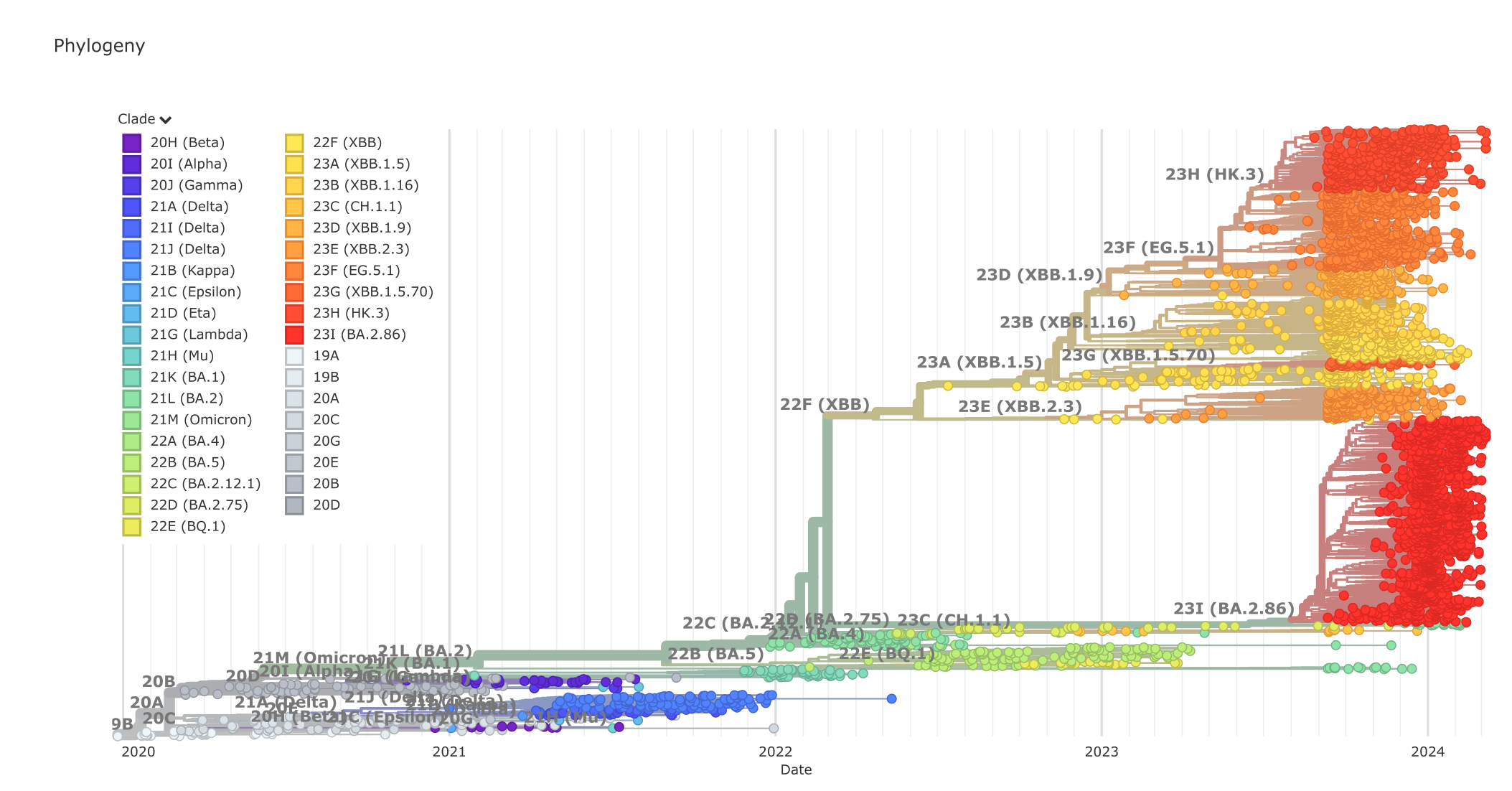
SARS-CoV-2 系统发育树,2024 年 3 月。来源:CC BY 4.0 [@sars-cov-2_2018]
其他例子包括研究 Covid-19 和其他病原体爆发、研究分子进化以及在多重序列比对(MSA)中替换的积累。或者研究物种内群体历史、重建历史生物地理学:在所有这些情况下,拥有一棵准确的系统发育树至关重要,因为我们希望能够重建进化谱系(系统发育树中的分支)以及它们如何进化、变化、复制或灭绝。所谓准确,是指估计尽可能接近实际(历史)关系,而这是我们无法确切知道的。由于这些事件发生在过去,我们无法"证明"它们,但它们是(关于关系的)假说,我们只能"印证"(确认、寻找支持证据)。
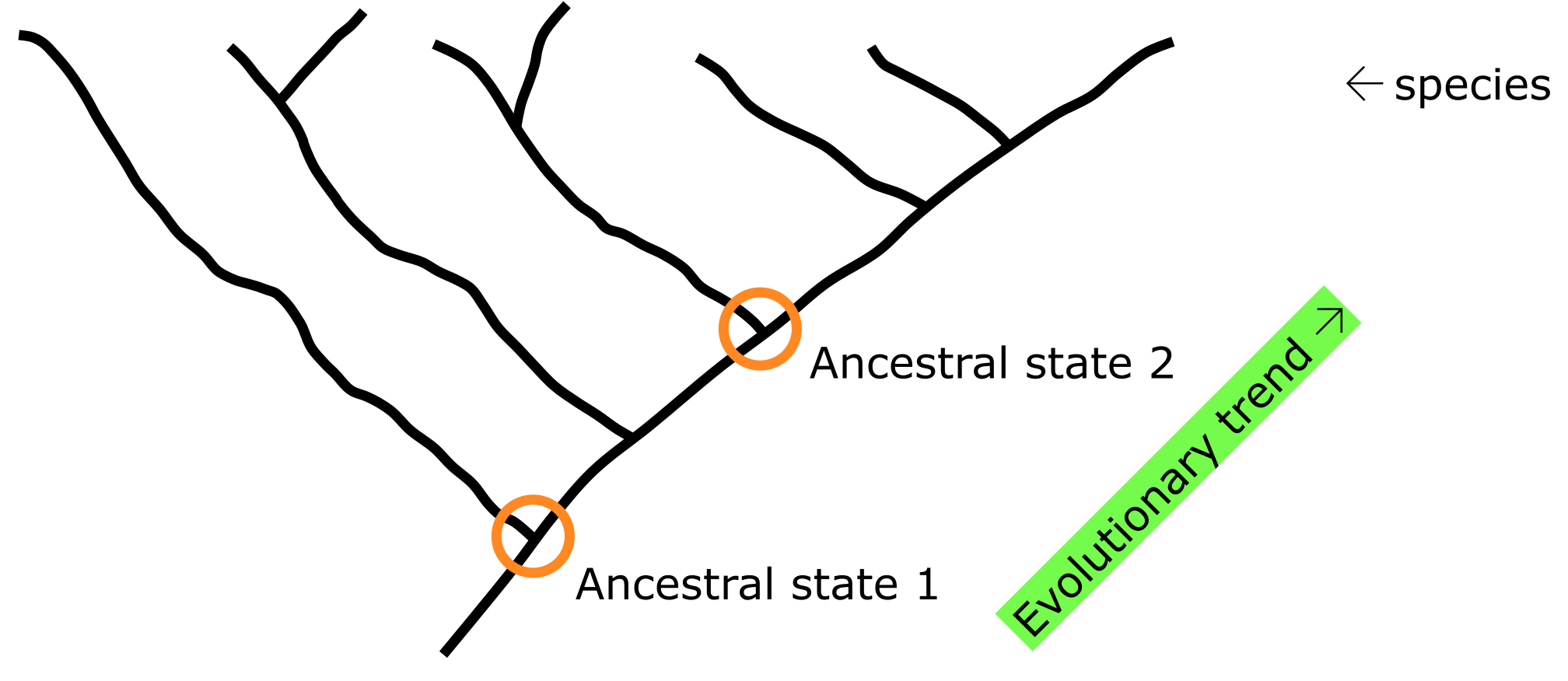
在系统发育树中比较物种(或基因)可以推断祖先状态和进化趋势。来源:CC BY-NC 4.0 [@own_3_2024]
当某个特定群体的系统发育树已知且已正确根化(Rooted)时,原则上可以重建其特征(Characters)在每个节点上的祖先状态(ancestral States)(例如蛋白质序列中的祖先氨基酸残基)。在此基础上,可以推断进化趋势(从历史到现状),从而研究特征的进化,即事物如何随时间变化。
系统发育树:结构与解读¶
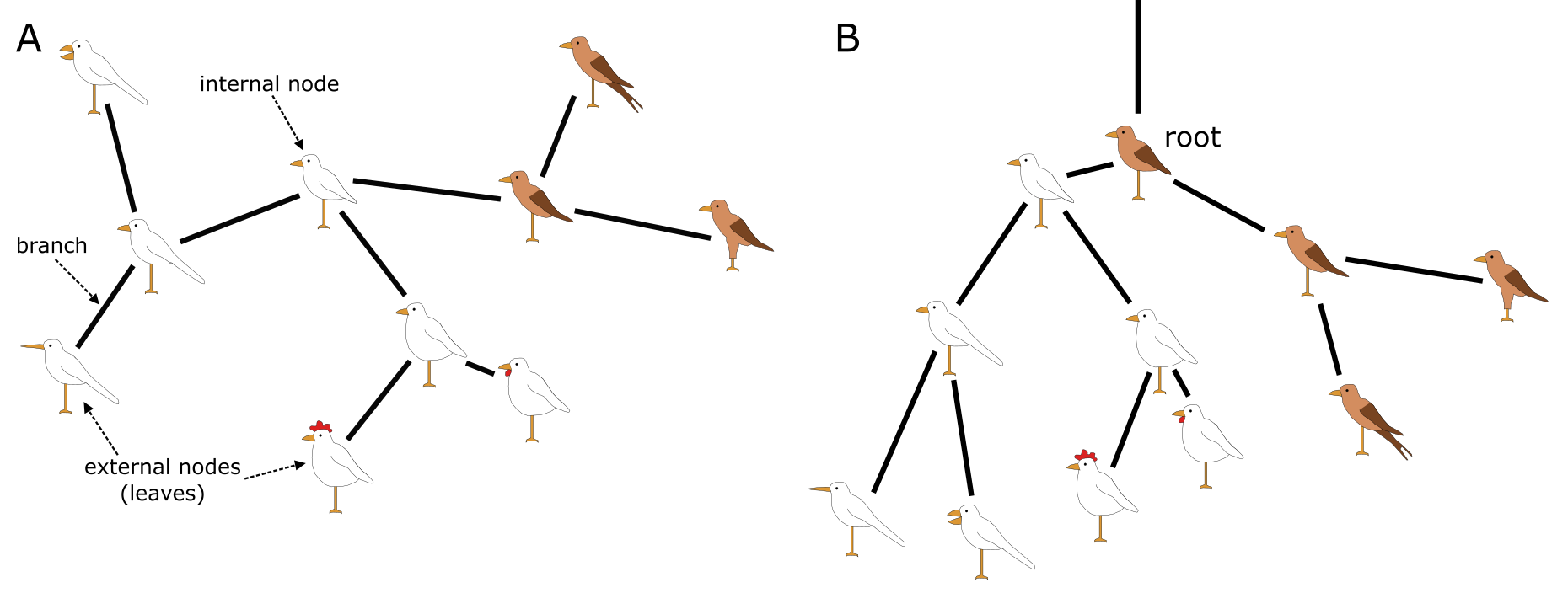
与所有树一样,系统发育树有主干、分支、叶片,理想情况下还有根。然而,系统发育树的特殊之处在于它们实际上是进化关系的假说,如上一节所述。叶片或外部节点是被观察和比较的个体(或序列),也称为操作分类单元(OTU,operational taxonomic units)或末端单元(terminals)。分支和节点是推断的谱系(lineages)或分支(clades),即未被观察到的。分支(Clade)是一个祖先节点及其所有后代,也称为单系群(monophyletic group),是单系性(Monophyly)的一个例子。它们通过系统发育树中连接 OTU 和 HTU(假想分类单元)的水平线来识别,如下面图所示。
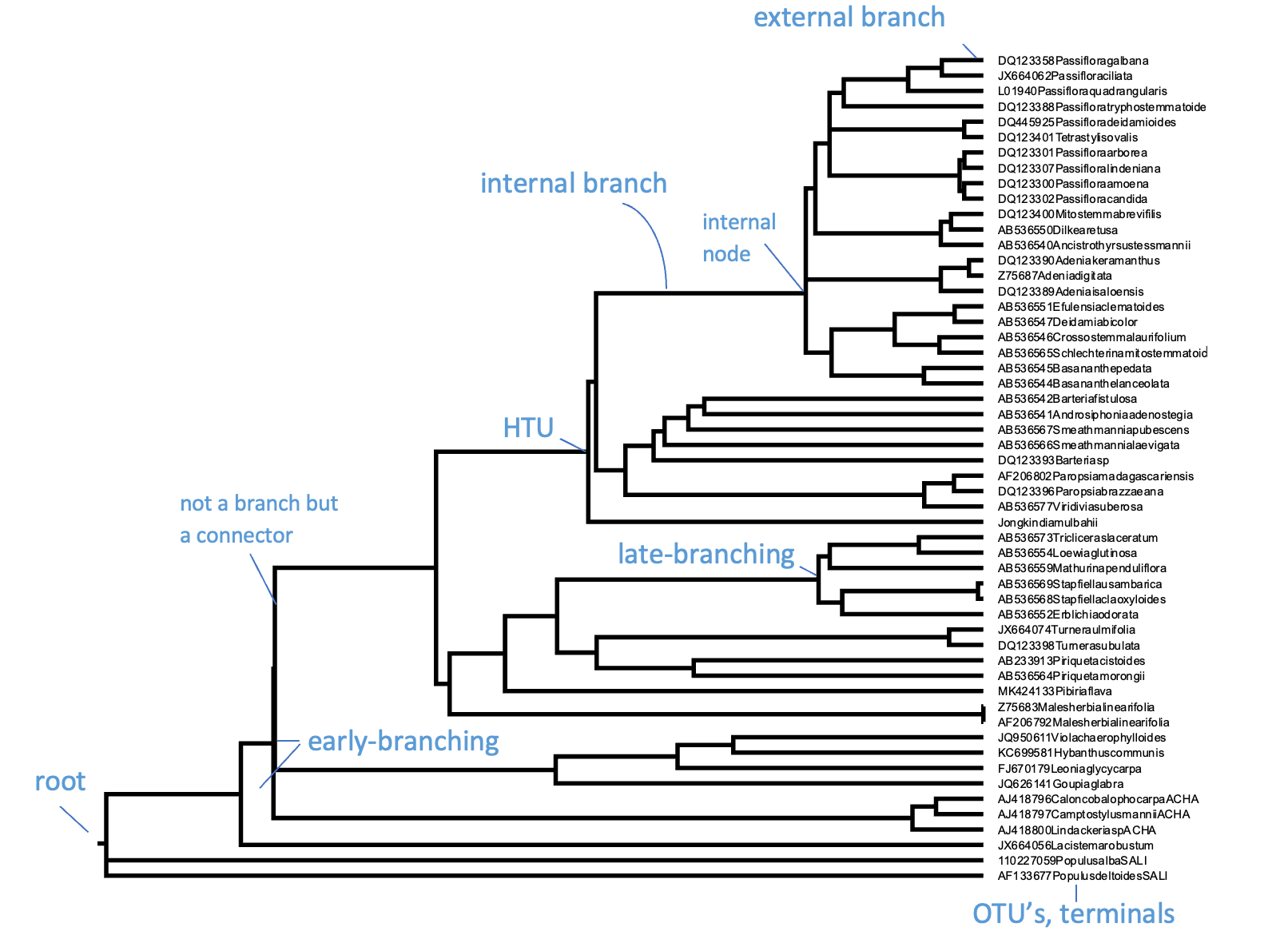
一棵有根的超度量系统发育树,标明了其主要部分和特征。此处,OTU 为 GenBank 植物叶绿体基因登录号,名称已缩写。来源:CC BY-NC 4.0 [@own_3_2024]
水平线代表实际的分支,而垂直线没有实际意义,仅用于连接分支和分支群;当包含更多末端单元时,垂直线会变长,但与数据(即 MSA)无关。分支通过节点连接,节点可以是内部的(HTU)或外部的(OTU)。内部节点代表未被观察或测序但被推断或重建的假想祖先。如上所述,外部节点是被观察的实际个体;它们从不直接相互连接,只通过内部节点连接。这些个体可以代表基因、物种或更高分类单元,但它们绝不能是类别(或平均值),因为特征和状态是对个体的不可分割的观测。分支和节点共同构成系统发育树的拓扑结构(Tree topology),即树的结构。
系统发育树最重要的方面之一是它是否有根,即我们能否区分哪些节点较老、哪些较新,以及存在哪些分支(clades)。根化(Rooting)通过选择一个外群(Outgroup)来完成,外群是感兴趣群体之外的一个参考分类单元(这在根化与分支一节中有更详细的描述)。需要注意的是,大多数系统发育重建方法实际上产生的是无根树,然后可以使用外群对其进行根化,以可视化进化的方向以及可以识别哪些分支。
相关与分歧¶
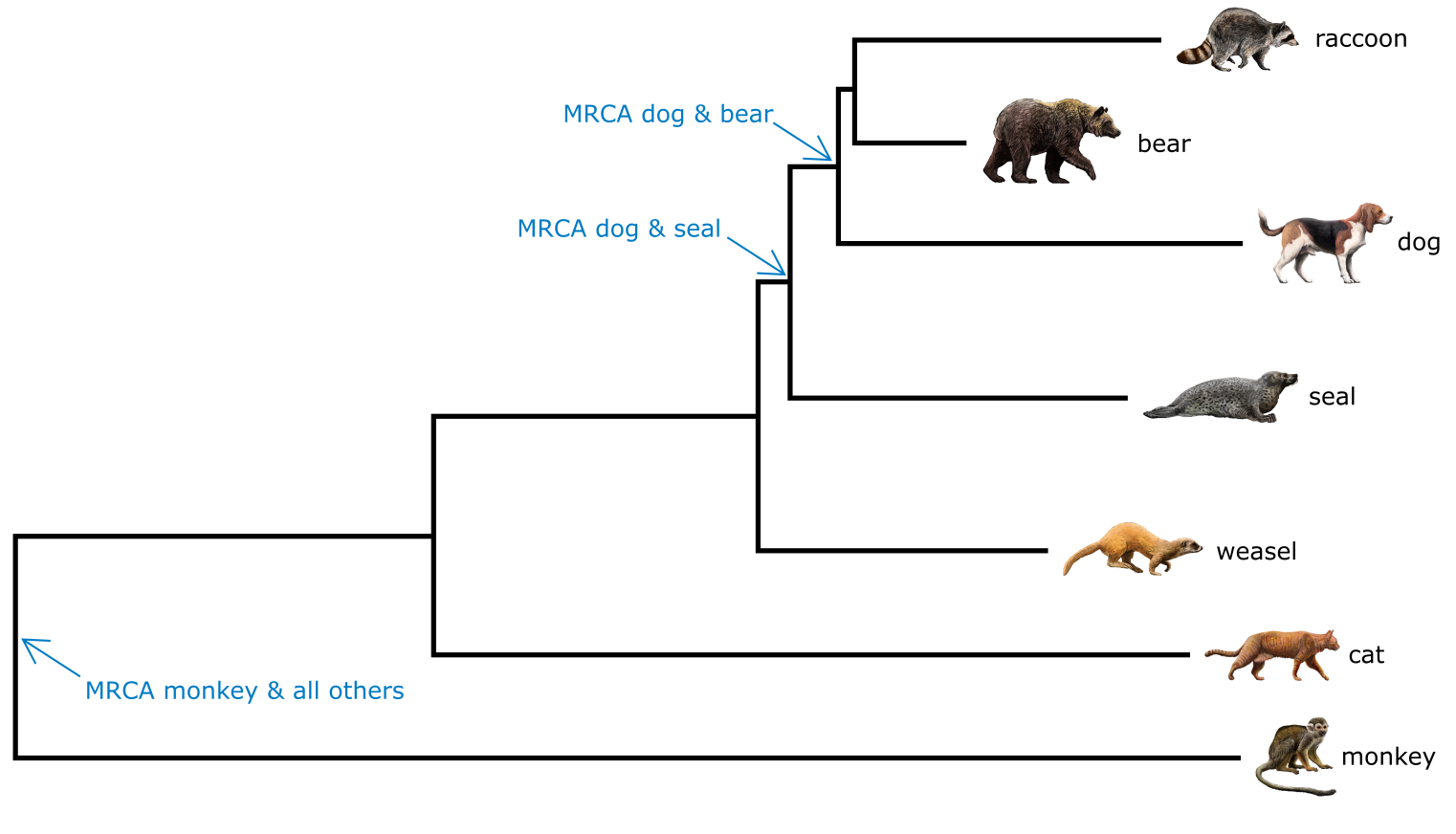
以猴子为根的哺乳动物加性系统发育树。猴、熊、海豹和狗的最近共同祖先已标出。树拓扑结构反映亲缘关系,分支长度对应分歧程度。来源:CC BY-NC 4.0 [@own_3_2024],图像素材来源:CC BY 4.0 [@DBCLS_2023]
在有根系统发育树中,共享较近共同祖先的末端单元比共享较远共同祖先的末端单元亲缘关系更近。因此,在上图中,狗和熊的亲缘关系比狗和海豹更近,因为狗和熊共享更近的共同祖先。另一方面,猴子和狗的亲缘关系与猴子和猫相同,因为它们都共享同一个最近共同祖先(MRCA)。不相关并不意味着分歧(diverged),分歧是指两条谱系自分裂以来积累的变化量,反映在分支长度(Branch length)中(或距离中)。在我们的例子中,浣熊和狗比浣熊和熊更分歧,但并不更近缘。
分支图、加性树与超度量树¶
系统发育树有三种类型:超度量树(Ultrametric tree)、加性树(Additive tree)和分支图(Cladogram)。当从根到每个外部节点的所有路径长度相等时,你可以将树的路径长度解释为与时间成正比,因此与其他路径同龄;节点的年龄原则上可以被推断。这种树称为超度量树,很容易通过其拓扑结构识别,其中所有末端分支对齐(通常向右)。另一种更常见的系统发育树是加性树,其中分支长度不与时间成正比,而是与数据集中变化量(即 MSA)成正比。因此,个体发生的变化(即替换、插入、缺失)越多,加性树中到其 MRCA 的分支就越长。大多数系统发育和建树平台或软件包产生加性树。生成超度量树通常需要额外步骤,每一步都引入不确定性。你可以说超度量树比加性树"距离数据远了一步"。只有当数据以严格时钟式方式积累替换(即像放射性衰变)时,超度量树和加性树版本才会相同。然而,这种严格的分子钟在实际数据中从未遇到过。最后,分支图(cladogram)样式的树是一种"示意图",仅用于显示树的拓扑结构。因此它具有人工的(相等的)分支长度,包括末端分支。
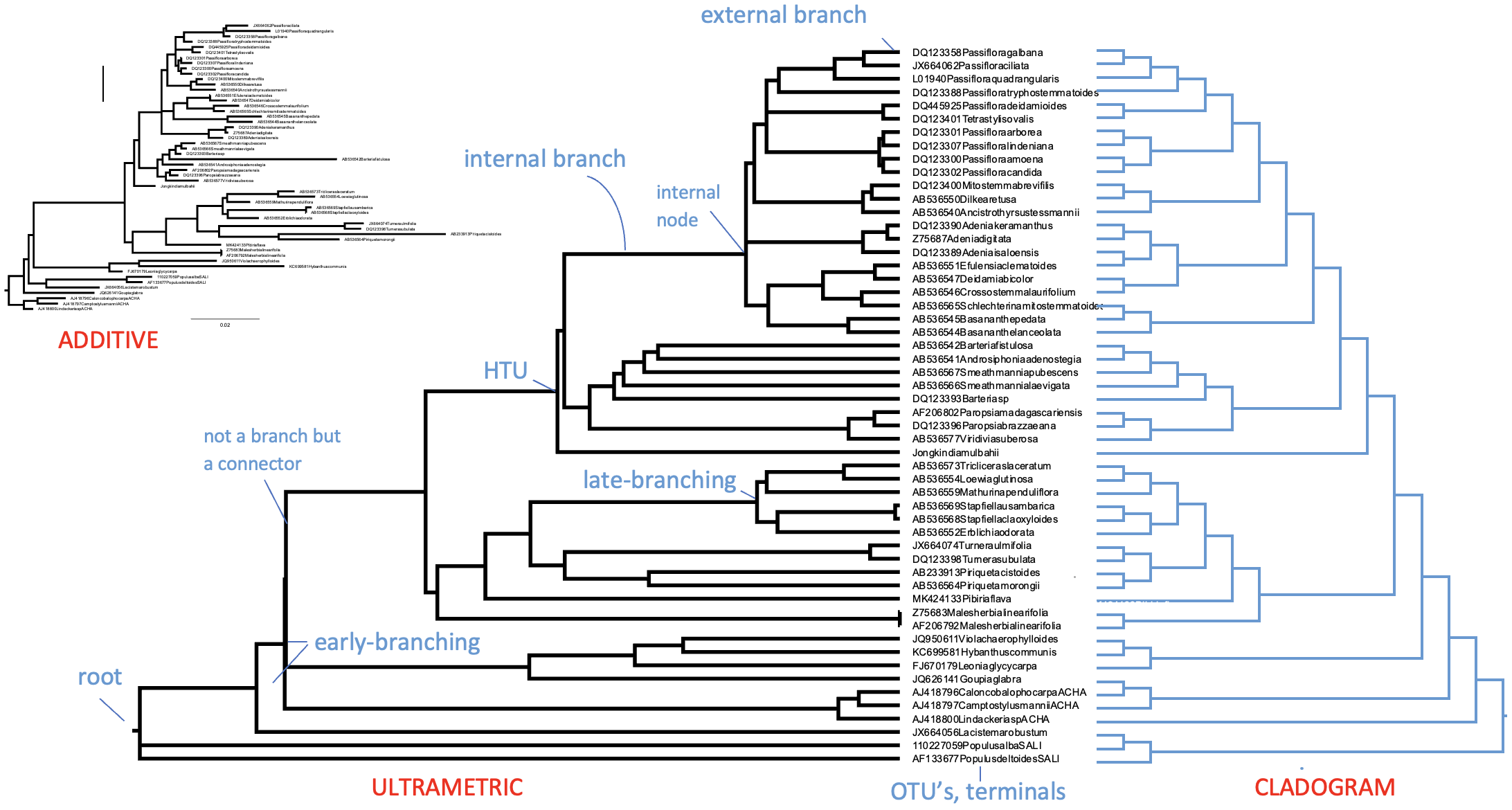
一棵有根系统发育树,标明了其主要部分和特征。此处,OTU 为 GenBank 植物叶绿体基因登录号,名称已缩写。对于相同数据,树以加性树(上)和超度量树(左下)形式呈现,分支长度对应时间。右图为翻转后的分支图,分支长度仅表示树的结构。来源:CC BY-NC 4.0 [@own_3_2024]
树的分辨率¶
系统发育树的分辨率是指从树中可以推断/观察到的节点和分支(分支群)的程度。树可以完全解析(fully resolved),在这种情况下每个内部节点连接三个分支:祖先分支和两个子分支。这样的树称为二叉树(bi-furcating)或二分树(dichotomous),意味着每个分支分裂为两个,在分支顺序或节点解析上没有不确定性。然而,系统发育树通常是部分解析的,包含多分支(Polytomies),即连接超过三个分支的节点。多分支代表系统发育树中谱系分支顺序不确定的部分。这可能是由于 MSA 中信息不足以解析谱系,或者是信息充足但信号冲突。多分支通常被解释为软多分支(soft),即使用的数据不足以解析推断的谱系。
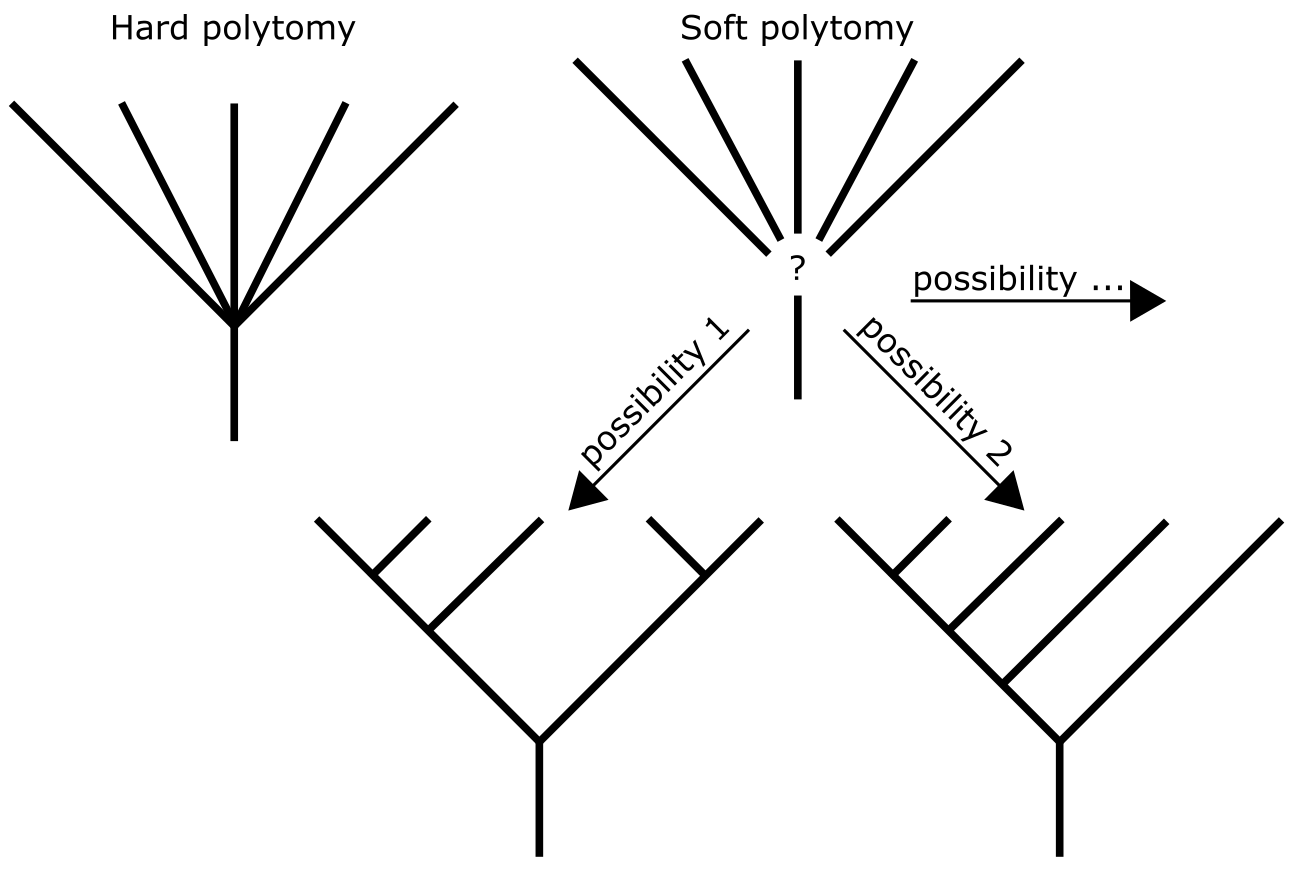
系统发育树中的硬多分支和软多分支。软多分支可能暗示不同的树分辨率。来源:CC BY-NC 4.0 [@own_3_2024]
相比之下,硬(hard)解释是:瞬时物种形成,即祖先物种谱系分裂太快,导致新谱系没有足够独特的替换来"标记"它们并在树中分配内部节点。一个例子是新生代晚期哺乳动物目的辐射,在地球两极周围较冷水域和热带的相对快速建立之后。几篇已发表的哺乳动物系统发育树包含未解析的脊柱或主干。
另一方面,基因树(gene trees,用于研究基因家族)也可能包含多分支,它们通常被视为软多分支(数据不够决定性以推断分支顺序)。全基因组复制(自体多倍化)在开花植物进化中相当常见。在这种事件之后,成对的基因可以预期是瞬间形成的,即没有积累独特的替换,可能导致基因树中的硬多分支。
直系同源与旁系同源¶
当末端单元实际上是基因或蛋白质序列时,树将是基因树(Gene tree),可能包含同源基因(homologs,源于共同祖先基因),也可能包含直系同源(orthologs)和旁系同源(paralogs)。直系同源性(Orthology)是由于物种形成事件而在谱系中出现的对应、同源(且大多相似)的基因。例如,人类 beta 珠蛋白和黑猩猩 beta 珠蛋白是直系同源物。通常,这些基因在不同物种中具有相同的功能,但并不一定如此。相比之下,旁系同源性(Paralogy)是由基因复制(Gene duplication)而非物种形成事件产生的相似基因。例如,同一物种中具有不同功能的基因家族中的蛋白质。这些相似基因称为旁系同源物(paralogs),在树上可视化为特定末端单元的多次出现。
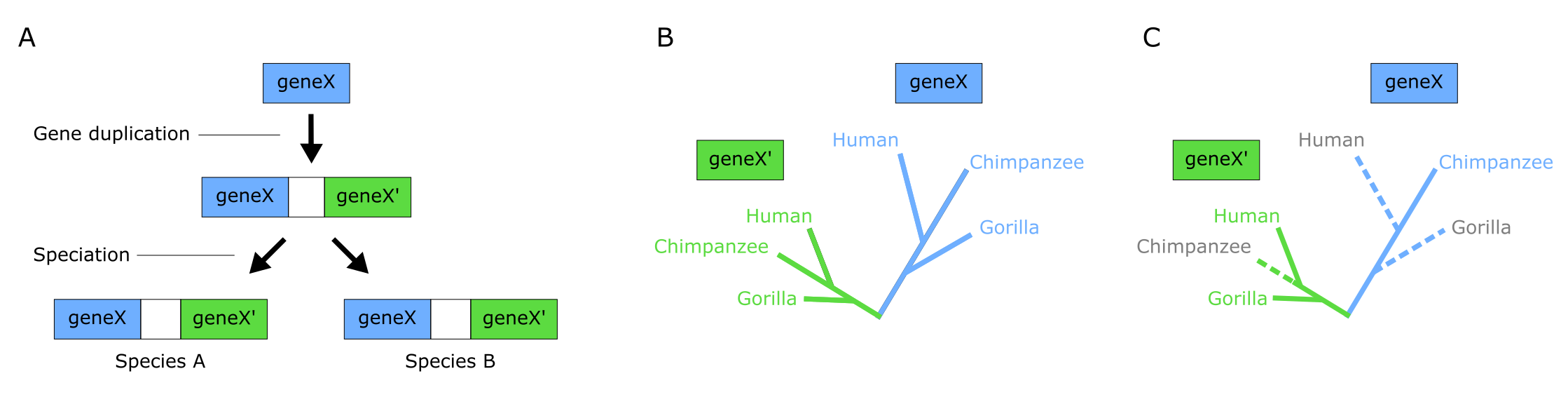
旁系同源的挑战:(A) 旁系同源基因由基因复制事件产生。基因 X 在物种 A 和 B 的最近共同祖先(RCA)中复制,产生旁系同源基因 X 和 X'。物种 A 和 B 继承了两个基因拷贝(除非其中之一在某处丢失)。(B) X/X' 基因家族的系统发育分析产生两个平行的系统发育树。基因 X 的所有序列互为直系同源物,基因 X' 的所有序列也是如此。但 X 和 X' 是旁系同源物。X 和 X' 子树都显示了三个物种之间的真实关系。子树也是彼此的自然外群,因此每个子树以另一个为根(互为根化)。(C) 如果不包含所有序列(由于不完整采样或基因丢失),X/X' 基因家族的树可能具有误导性。如果虚线分支缺失,则真实的物种关系会被错误表达。来源:CC BY-NC 4.0 [@own_3_2024]
在下图中,展示了一系列涉及两次复制和一次物种形成事件的过程,可以导致两个物种中一组同源基因。其中一些是直系同源物,另一些是获得了新功能的旁系同源物。物种树由浅蓝色圆柱体描绘,圆柱体中的分支点(节点)代表物种形成事件。在祖先物种(顶部)中,一个基因以单拷贝形式存在并具有功能 α(蓝色)。一段时间后,基因组内发生基因复制事件,产生两个相同的基因拷贝,其中一个随后进化出不同的功能,标记为 β(红色)。因此,α 和 β 现在是旁系同源基因。后来,发生物种形成事件,产生两个物种(A 和 B),都含有基因 α 和 β。物种 B 中的基因 Bα 随后经历另一次复制事件,产生旁系同源基因 Bα 和 Bγ。经过进一步的分歧进化后,Bγ 获得新功能 γ(绿色)。Bα 基因在功能上仍与原始基因 α 非常相似。在进化期末,两个物种中的所有五个基因都是同源的,具有三对直系同源对:Aβ/Bβ、Aα/Bα 和 Aα/Bγ。Bα 和 Bγ 是旁系同源物,除直系同源对外的所有其他组合也是如此。注意 Aα 和 Bγ 尽管功能不同,但它们是直系同源物。
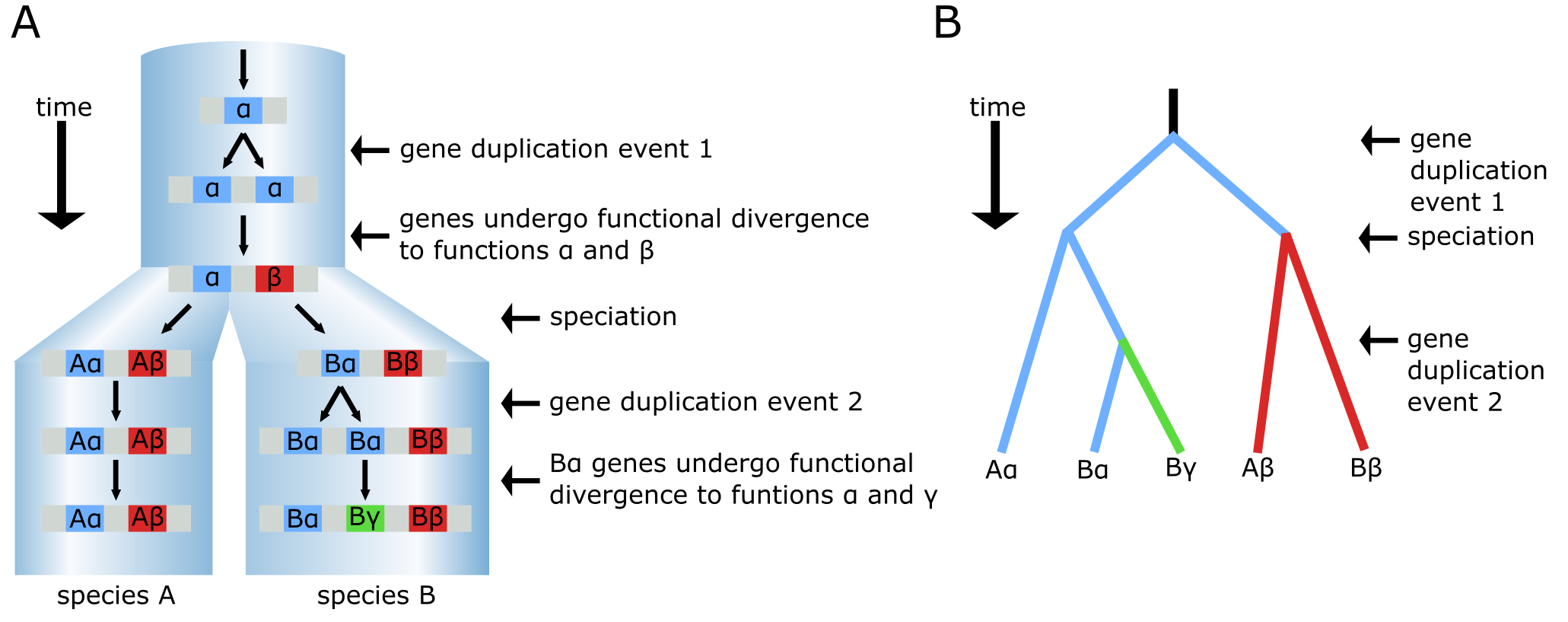
一个经历了两次独立复制事件的基因的进化历史。(A) 物种树(大蓝色圆柱体)包含物种 A 和 B,标明了导致 β 和 γ 功能的基因复制和新功能化事件。(B) 基于 (A) 中产生的 5 个基因绘制的系统发育树,此处以分支图形式呈现。来源:CC BY-NC 4.0 [@own_3_2024]
关于基因树与物种树的另一个例子,考虑基于白细胞介素基因序列比较的树。物种树中的末端单元(牛、羊、猪等)在基因树中多次出现,每种与不同的白细胞介素序列类型分组在一起。这些是旁系同源物,可能是在白细胞介素分支增殖过程中基因复制事件产生的。
鉴于人类中有四个 IL1 拷贝(在基因树中以绿色方框标出),白细胞介素的历史中至少发生了三次基因复制。其中三次是从同一哺乳动物物种中存在多个 IL 拷贝推断出来的,即 i) 在 IL-1α + β 分支的祖先中,ii) 在 IL-1rα + β 分支的祖先中,iii) 在 IL-1β 分支中作为姐妹对。复制 δ3 是解释哺乳动物物种树与 IL-1β 系统发育之间不一致性所需要的。这种不一致性是在基因树中人类和小鼠的亲缘关系比它们与牛/羊的亲缘关系更近。然而,物种树表明人类与牛/羊的亲缘关系比与小鼠更近。为解决(协调)这一问题,δ3 被建议如下所示。
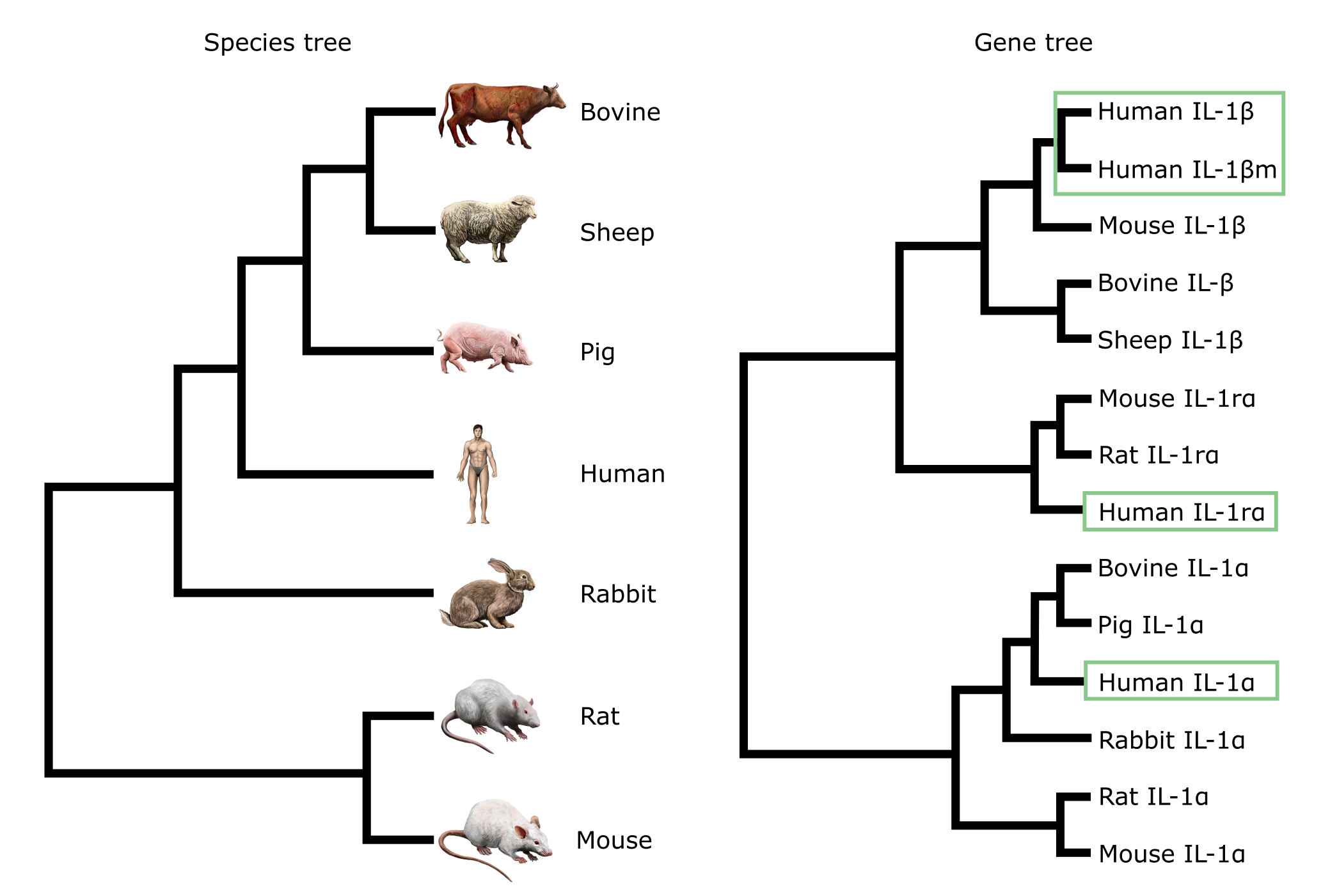
基于外部证据的物种树(左)和基于哺乳动物白细胞介素-1 基因比较的基因树(右)。在基因树中,可以看到 alpha 和 beta 拷贝,它们可能是由基因复制产生的。基因树中"人类"的出现以绿色方框标出。来源:CC BY-NC 4.0 [@own_3_2024],图像素材来源:CC BY 4.0 [@DBCLS_2021]
下图中的树是所谓的协调树(reconciled tree),它被推断为一棵扩展树,需要假设这棵扩展树才能解释基因树中所有白细胞介素序列类型的位置和分布。除了四个基因复制(标记为 δ1、δ2、δ3 和 δ4)外,还需要假设几个基因丢失(以浅灰色分支标出)才能解释基因树中的模式。
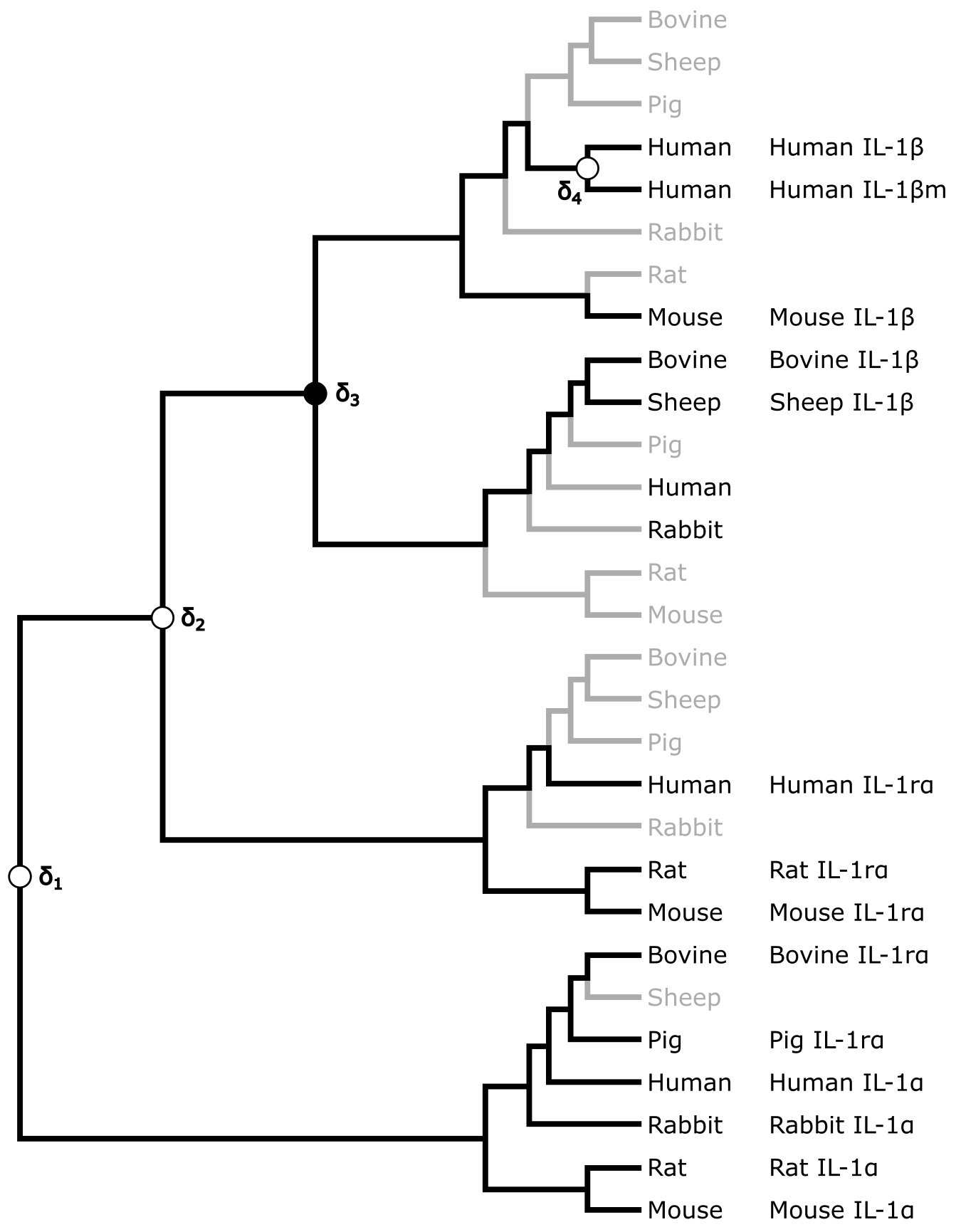
上图所示的哺乳动物白细胞介素-1 基因树的协调树。基因丢失以浅灰色标出。在所需的四次复制中,三次由同一哺乳动物物种中存在多个 IL 拷贝支持,一次(δ3)需要解释 IL-1 与哺乳动物系统发育之间的不一致性。来源:CC BY-NC 4.0 [@own_3_2024]
框 3.1:物种树估计分析
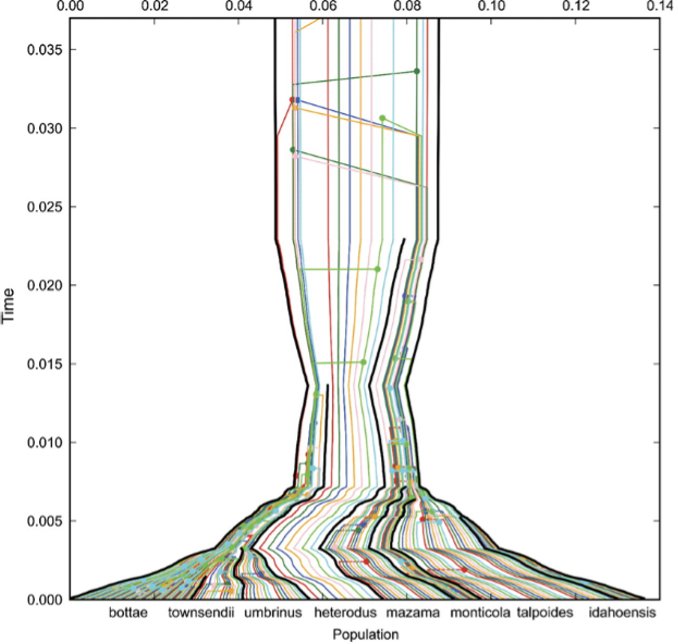
基因树(彩色)嵌入西部囊鼠(Geomyidae, Thomomys)的物种树(黑色线)中。来源:CC BY-NC 2.5 [@embedded_tree_2009]
当末端单元是旨在代表物种的个体时,我们原则上是在推断物种树(species tree)。上图展示了多个基因树(彩色)通过物种树估计分析(species tree estimation analysis)对物种树(以黑色线标出)的贡献。不同的物种名称,此处为 Thomomys "西部囊鼠"(Geomyidae),沿水平轴标出,它们的聚合(coalescence,基因谱系向后回溯合并)可以随时间追踪。此类分析超出了本课程的范围,但始终牢记你的系统发育重建处于什么水平——物种水平、基因水平还是生物地理区域水平——很重要。
系统发育树中的节点支持度:Bootstrap 方法¶
系统发育树并非所有部分都同等有数据(MSA)支持。我们估计的节点周围可能存在相当大的不确定性,影响我们对这些节点的信心。在实验科学中,通常使用某种统计量来量化不确定性,例如重复实验结果的均值和标准差。然而,系统发育学不是实验性的,而是寻求重建由进化驱动/塑造的历史模式。如本章开头所述,这意味着我们无法"证明"系统发育,也无法重复它们,甚至无法知道我们是否重建了正确的系统发育。我们"只是"将系统发育树作为真实系统发育(true phylogeny)的估计。
为了衡量系统发育树中的节点支持度(Nodal support),与其生成多个 MSA 的重复(很可能全部相同),我们可以从 MSA 中抽取随机样本,并使用这些伪重复(pseudo-replicate)数据集来建树。多次重复此过程(数百或数千次)并总结由此重建的树之间的变异,可以深入了解数据结构及其如何支持树中的节点。这实际上衡量的是系统发育估计的采样方差。这个过程称为 Bootstrap 分析,将在最大似然建树一节中进一步讨论。
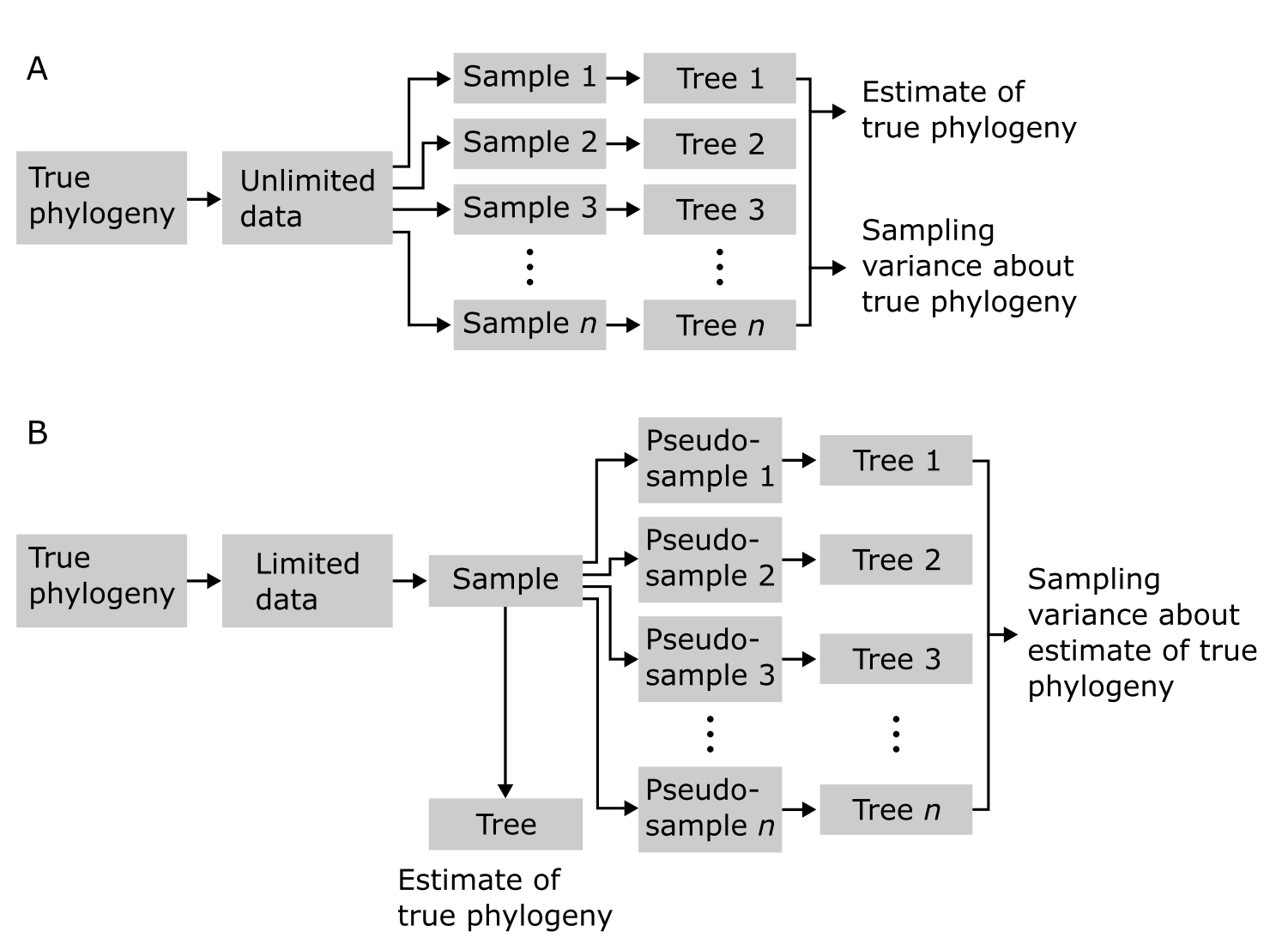
系统发育重建中的 Bootstrap 重抽样分析。在无限数据(A,不现实)情况下,基于样本的树的汇总产生真实系统发育的采样方差。在有限数据(B,现实)情况下,只有伪样本可用,汇总系统发育估计的采样方差。来源:CC BY-NC 4.0 [@own_3_2024]
特征与系统发育树¶
如上所述,系统发育树不是直接观察到的,而是推断出来的,代表进化关系的假说,基于共享的历史对个体进行分组。用于比较的数据是在 MSA 中对齐的同源位点(氨基酸或核苷酸),其中替换是可见的。此处的同源(Homologous)是指基因序列中由于共同祖先而产生的对应位置。因此,例如一个物种基因序列中的位置 423 与另一物种基因序列中的位置 423 是同源的,因为两个物种共享最近的共同祖先,我们假设这些基因是直系同源物。每个这样的位置被视为一个具有状态的特征,在 MSA 中高效可视化为替换或树上的"步"(steps)。MSA 中的所有核苷酸或氨基酸替换,包括唯一替换(仅在单个个体或序列中出现)和共享替换(在至少两个序列中出现),都用于构建系统发育树。然而,不变特征(不显示替换)预期不会对建树过程有所贡献,因为它们不包含比较信号。共享替换对于构建树的分支有信息量,因为它们将末端单元分组并增加内部分支长度。唯一替换仅贡献于"细枝"或外部分支长度,没有分组能力。MSA 中一组序列的共享替换越多,系统发育树中结果节点的支持度就越高。
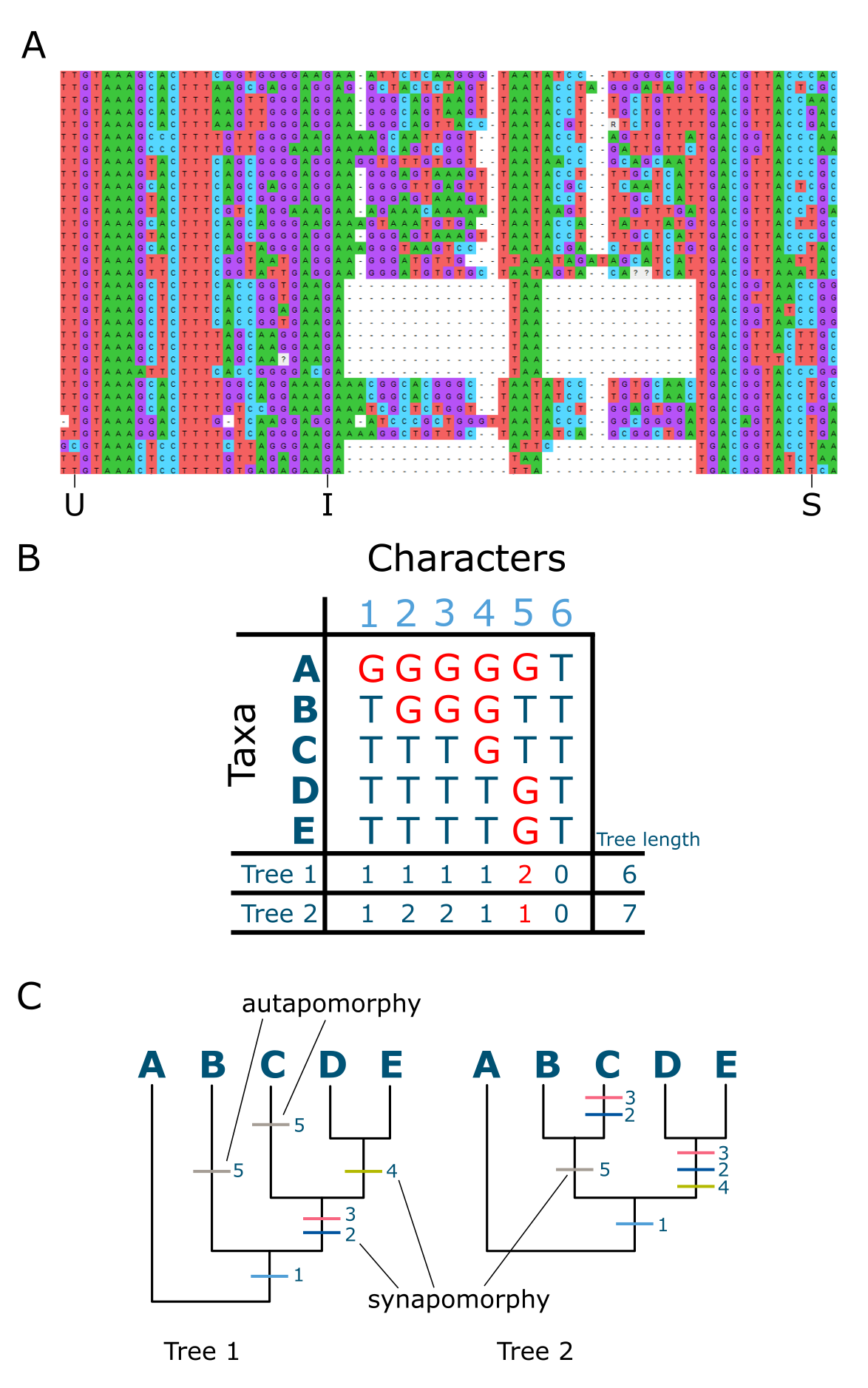
特征与系统发育树。A:多重序列比对(MSA,核苷酸),标出了共享衍征(S)、唯一衍征(U)和不变特征(I)的示例;B:包含 S、U 和 I 特征的 MSA 以及每个特征的步数,tree1 和 tree2 的总树长标在底部;C:两棵"候选树"作为解释 MSA 数据的替代假说,所有特征的状态变化标在树上,示例共同衍征和自衍征已标出。注意特征 6 是不变的,因此不对任何树有贡献。树 1 比树 2 少一步,因此是优选树。来源:(A) 使用 MEGA11 创建并修改自相关文献 [@mega_2021] (B & C) CC BY-NC 4.0 [@own_3_2024]
在 MSA 中观察替换时,我们无法确定哪些是祖先状态(已在"深层"祖先中出现)哪些是衍征状态(更近期出现的)。但置于有根系统发育树的背景下,共享替换实际上是共享衍征替换,称为共同衍征(synapomorphies,syn = 共享,apo = 衍生,morphy = 特征),而唯一衍征替换称为自衍征(autapomorphies)。
在设计系统发育研究时(涉及一个或多个 MSA 的编制),通常需要在添加更多特征(加长 MSA)和添加更多末端单元(向 MSA 添加更多序列/行)之间做出选择。虽然前者很诱人,但通常添加末端单元(分类单元)更有用,因为这可以实现更多共同衍征。毕竟,共同衍征是相对的(非绝对的)实体:只有在其他序列的背景下才能真正"看到"它们。例如,当研究一个在谱系进化过程中发生过复制的基因家族时,应在 MSA 中包含许多分类单元以捕获复制事件。仅添加更多特征可能会放大分类抽样不足导致的错误或假象。这可能导致错误推断的长分支,其位置和节点看似具有高支持度。这种现象称为长枝吸引(long-branch attraction),将在估计序列分歧度一节中进一步讨论。
根化与分支¶
分支(Clade)是一个祖先节点及其所有后代,也称为单系群(monophyletic group)。我们通常将分支的祖先节点称为推断的最近共同祖先(MRCA)。当然,总会存在较远("更深层")的祖先,但它们可能对识别和推断分支及其关系没有信息量,因为它们也是其他分支的祖先。在深层分歧处(如鲱鱼与果蝇),同源性和所用特征的解析可能不够清晰和充分。
系统发育树中的信息是层级性的,结构是其他更包容结构的组成部分。分支通常确实嵌套在一起,即一个分支是更大分支的子集。除了嵌套外,分支还可以互为姐妹(sister groups),意味着它们共享一个独占的最近共同祖先(MRCA),且不包含其他分支。姐妹群(Sister group)在进化和比较研究中非常有用,因为它们代表完全同龄的谱系。
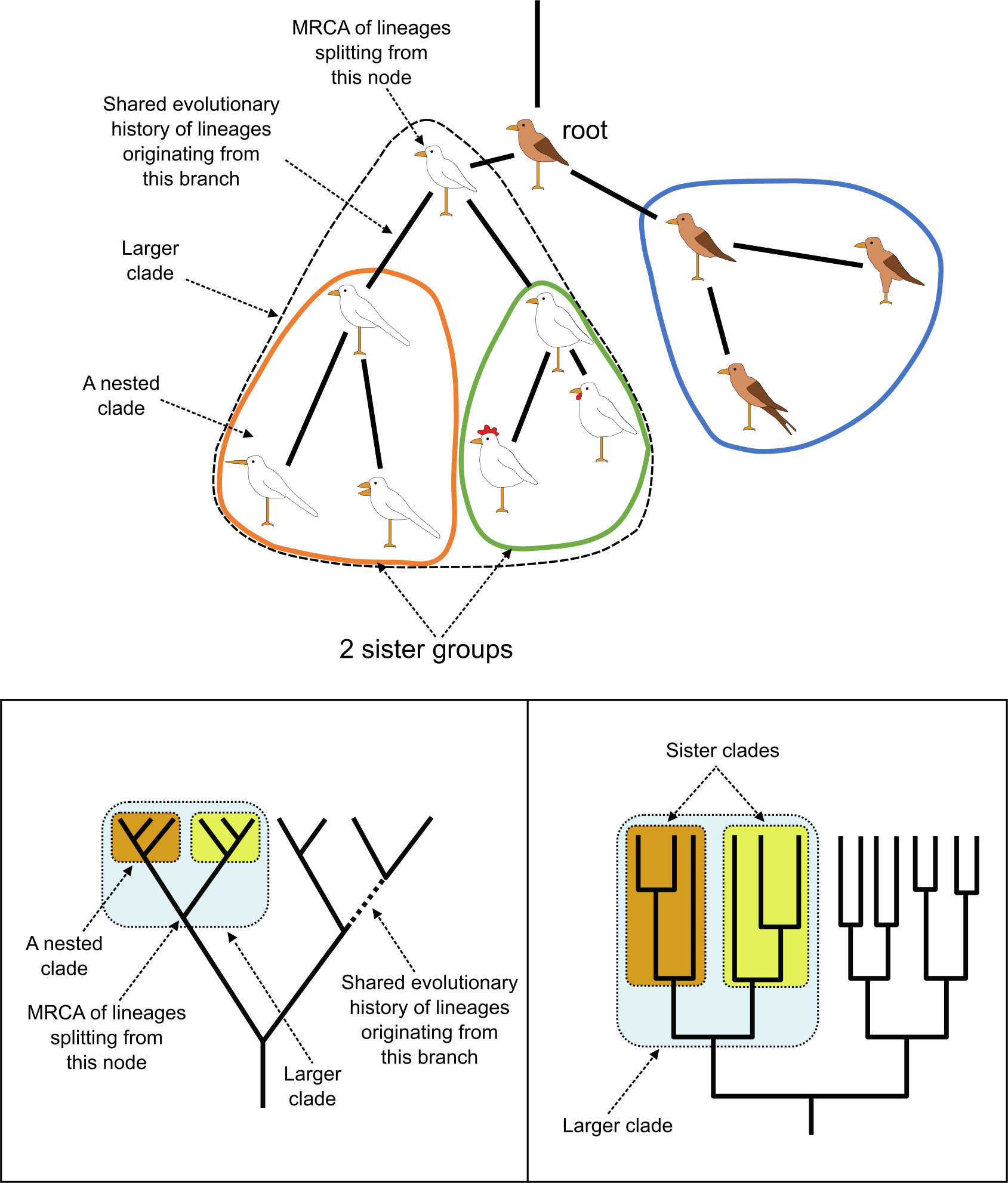
嵌套分支和姐妹分支。上图,与有根树相同,现标出了嵌套分支(橙色和绿色形状):小橙色分支嵌套在更大的虚线分支中;它也是绿色分支的姐妹分支;蓝色分支和更大的虚线分支也是如此。下图,嵌套和姐妹分支,标出了 MRCA。来源:CC BY-NC 4.0 [@own_3_2024]
再次强调,MRCA 及其所有后代被认为构成一个分支。这个分支可以成为进一步分析或分类的基础。需要认识到的是,分支基于观察(共同衍征),因此代表证据,而分类本质上是主观的(意见),是对分支的解读和使用。例如,当分支的某个后代在分类中被遗漏,例如脊椎动物从无脊椎动物中被遗漏,或鸟类从恐龙中被遗漏时,所提出的分类单元或分类不再是单系的,被视为并系群(paraphyletic group),即 MRCA 并非其所有已知后代,这是并系性(Paraphyly)的一个例子。并系群(也称为非自然群)仍在使用,但不被认为是分类的适当基础。
在研究基因家族及其进化时,比较基因树中各分支(尤其是姐妹分支)是有用的,因为它们完全同龄。它们之间的任何差异——替换率、序列组成偏差或每个分支的谱系数量——都是由于物种形成过程造成的,而非分支年龄差异。为了进行比较,重要的是比较单系群而非并系群,因为后者不能直接比较或同龄。
根化(Rooting)树是将其极化,区分老节点和年轻节点。当树被正确根化(通常使用感兴趣群体之外的外群参考分类单元)时,它在祖源方面是有方向的,因此可以推断分支(注意无根树是非极化的,原则上不包含分支而是"氏族"或"潜在分支")。在上图的 B 中,我们看到鸟类系统发育树的有根版本似乎多了一个棕色鸟类。外部证据(图中未显示,只有顶部通向外群的垂直分支)显然足以将根放在棕色鸟类和白色鸟类之间。因此,一个新的内部棕色鸟类被推断为 MRCA,外群分支可以连接到它。然而,这使得棕色鸟类相对于白色鸟类是并系的,因为并非棕色 MRCA 的所有后代都是棕色的,有些是白色的。白色鸟类现在是单系的。
系统发育树的根化。(A) 描绘一组白色和棕色鸟类物种系统发育关系的无根树;外部节点代表现存(活的、被观察的)物种,各自具有形态学的共同衍征或自衍征;内部节点代表推断的(未被观察的)祖先。树是完全解析的,每个内部节点连接三个分支。看相邻内部节点上的棕色和白色鸟类,不清楚进化的方向,是棕色产生白色还是反过来。这可以通过根化树来实现,通常基于与外部参考物种的比较。(B) 有根树;外部证据(未显示)显然足以将根放在棕色和白色鸟类之间。因此,一个新的内部棕色鸟类被推断为 MRCA,使棕色鸟类相对于白色鸟类成为并系群,而白色鸟类现在是单系的。来源:CC BY-NC 4.0 [@own_3_2024]
不当的根化会影响分支和树的整体结构(如下图所示)。此树的正确根化已标出,人和黑猩猩为姐妹群,这是基于这些物种的外部证据而无可争议的。将根放在无根树五个末端单元的七个可能不同位置上,只有三种情况下保持正确的人-黑猩猩-大猩猩单系性。其他四种拓扑结构显示出广泛的冲突,彼此之间以及与正确拓扑结构之间都不一致。这表明在选择和分配合适外群时应谨慎,在孤立的系统发育长分支(如原生生物或浮游动物谱系)或重建基因树时可能有问题。在这种情况下,通常考虑在远缘进化谱系中(如被子植物的 Amborella)的感兴趣基因的足够相似的拷贝作为根化该基因树的合适外群。下图展示了一棵带加性分支长度的无根树示例。
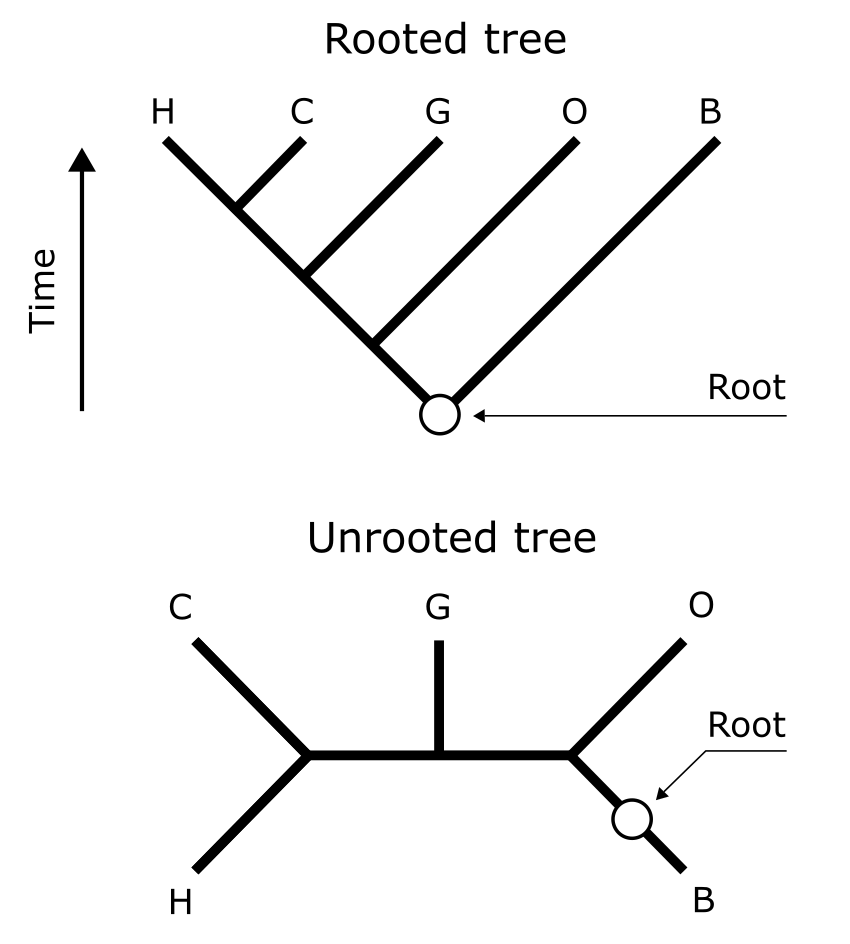
系统发育树的根化。标出了人(H)、黑猩猩(C)、大猩猩(G)、猩猩(O)和长臂猿(B),有根树(上)表示基于外部证据的正确树拓扑结构。此根的位置在有根树和无根树中标出。来源:CC BY-NC 4.0 [@own_3_2024]
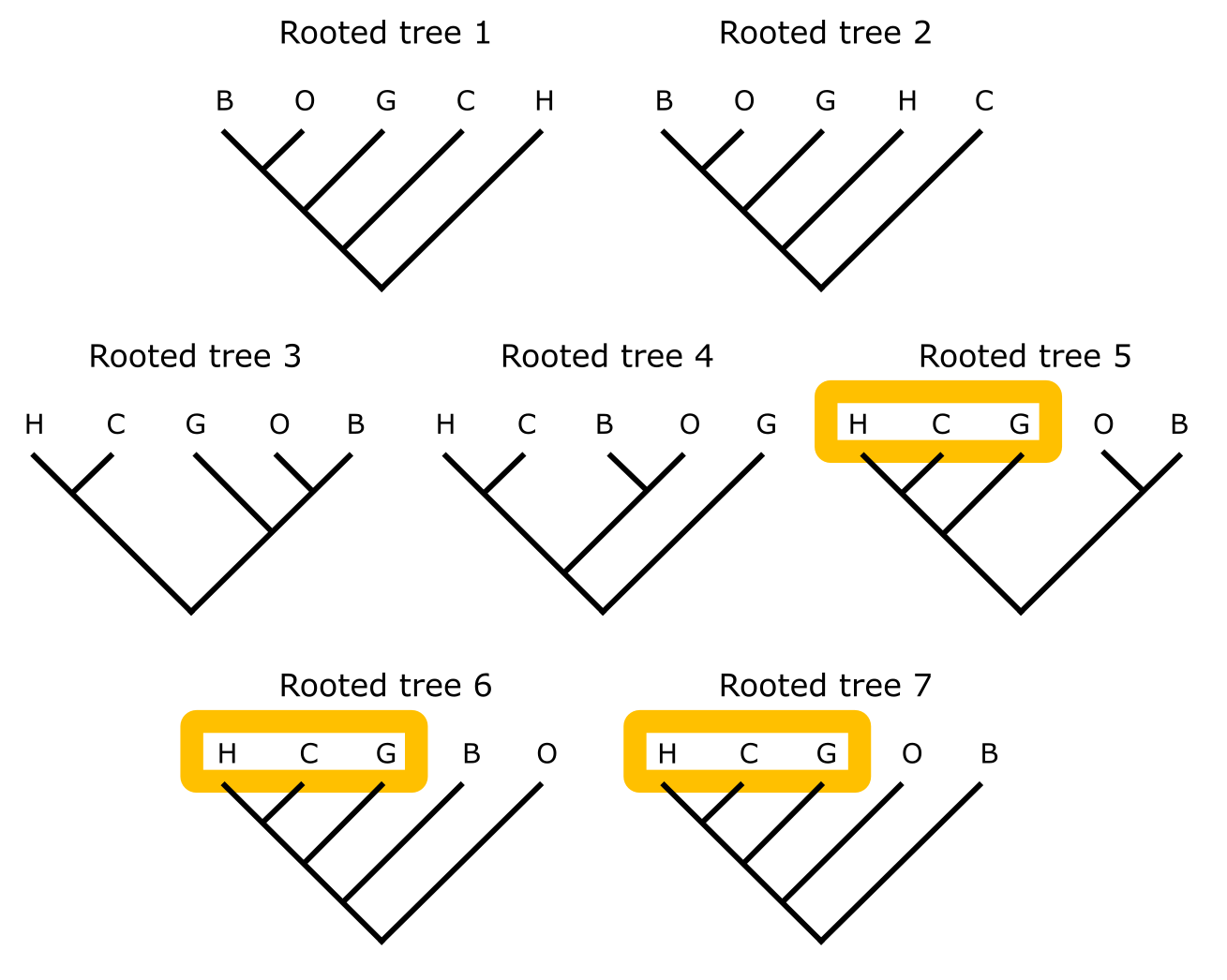
系统发育树的根化。从上图中五个序列的无根树可以推导出的七棵有根树。每棵有根树 1-7 对应将根放在无根树的不同分支上。末端标签与上图相同;橙色形状表示人、黑猩猩和大猩猩的单系性(当存在时)。来源:CC BY-NC 4.0 [@own_3_2024]
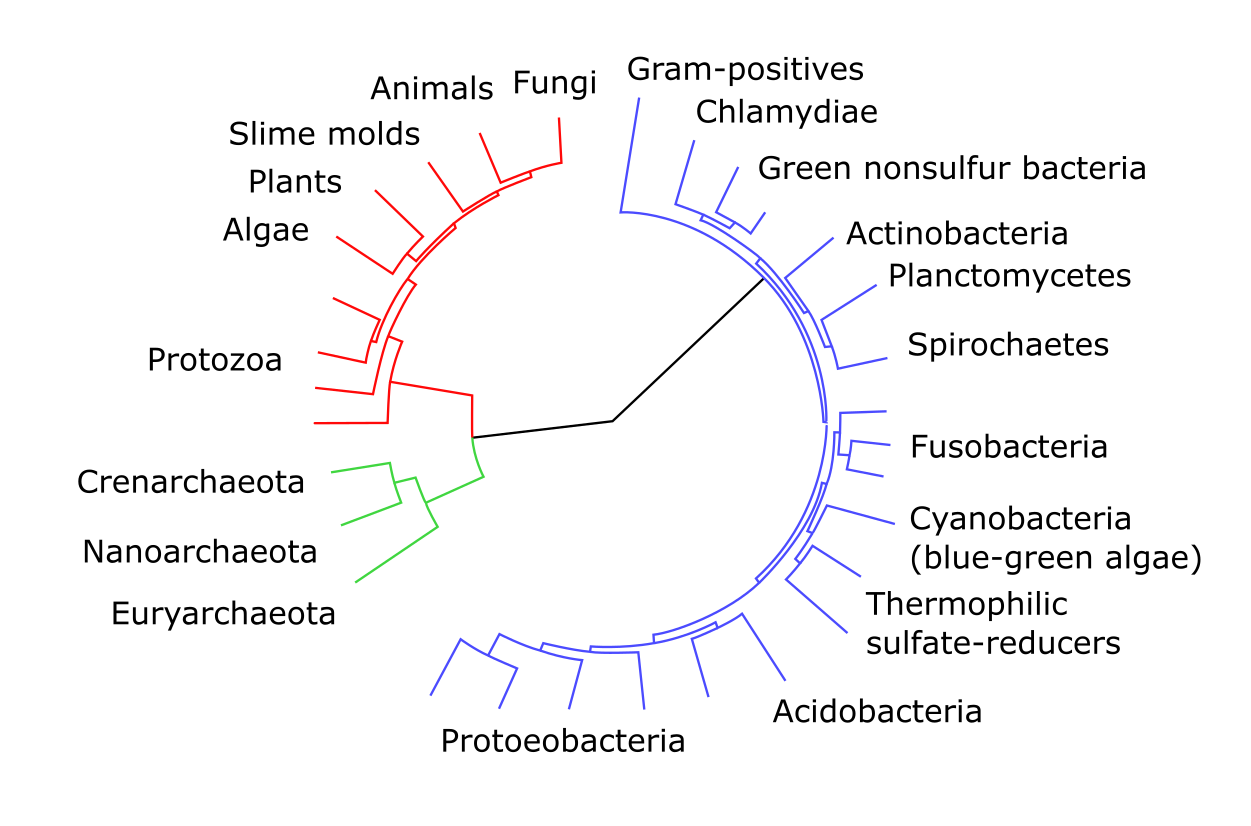
包含古菌、细菌和真核菌群的无根树示例。颜色标记表示可能构成氏族的群体,取决于树如何被根化。来源:CC0 1.0 [@unrooted_tree_2009]
Newick 树表示法¶
系统发育树是图形结构("图"),是系统发育重建(有时涉及数百或数千个序列)的结果,特别是在使用基于特征的树搜索时(见下文建树的主要方法),可能存在大量"最优树",都需要加以考虑,例如通过计算一致树(consensus tree,见树空间与启发式搜索方法)。无论如何,在系统发育和生物信息学分析流程中处理大量树需要树图以一种易于读取和生成的格式表示,即线性语句。为此,常用 Newick 格式,其中括号描述树的结构。例如,上图中的有根树在 Newick 格式中看起来像 ((((H,C)G)O)B)。如果树有分支长度,也可以在此格式中标注(另见 Brightspace 上建议的 Newick 树练习)。
框 3.2:Newick 格式树重建
(Photobacterium_profundum:0.0713485869,Vibrio_cholerae:0.0572857616,(((((Coxiella_burnetii:0.0942829505,(((((Bartonella_henselae:0.0149660313,Bartonella_quintana:0.0104976443)100:0.0417236852,(Bradyrhizobium_japonicum:0.0661124319,Brucella_melitensis:0.0305911464)84:0.0159603838)100:0.0717972517,Caulobacter_crescentus_CB15:0.1317093492)89:0.0321405560,((Rickettsia_prowazekii:0.0221103197,Wolbachia_pipientis:0.0257883738)76:0.0009060796,Rickettsia_conorii:0.0023047443)100:0.1908928012)100:0.1212044773,((Campylobacter_jejuni:0.1640483645,(Helicobacter_pylori:0.0777846753,Helicobacter_hepaticus:0.0262963532)97:0.0750887828)100:0.3046033428,((Geobacter_sulfurreducens_PCA:0.0953158405,Desulfovibrio_vulgaris:0.2786904064)52:0.0512359438,Bdellovibrio_bacteriovorus:0.2548155296)65:0.0488493427)81:0.0360783687)100:0.0960879477)74:0.0330231208,((Xylella_fastidiosa_9a5c:0.0356264562,Xanthomonas_axonopodis_pv._citri:0.0296447972)100:0.0938726215,(((Bordetella_parapertussis:0.0000025222,Bordetella_pertussis:0.0033397334)100:0.0907958037,Ralstonia_solanacearum:0.0732876166)100:0.0326570294,(Chromobacterium_violaceum:0.0690355638,Neisseria_meningitidis:0.1027698727)75:0.0194002885)100:0.1171829439)81:0.0311798765)91:0.0328742881,(Acinetobacter_sp.:0.1353093927,(Pseudomonas_aeruginosa:0.0385742686,(Pseudomonas_putida:0.0073082084,Pseudomonas_syringae:0.0208918284)89:0.0075987307)100:0.0665927895)63:0.0151201359)100:0.0903890667,Shewanella_oneidensis_MR-1:0.0737971703)87:0.0414970304,((((Salmonella_typhimurium:0.0349235213,((Shigella_flexneri:0.0016182221,Photorhabdus_luminescens:0.0008262939)99:0.0086172615,Escherichia_coli_K12:0.0013793749)99:0.0106569199)99:0.0258934845,Yersinia_pestis:0.0509601305)88:0.0118616315,(Buchnera_aphidicola:0.1429434318,Wigglesworthia_glossinidia:0.1005118155)97:0.0480006109)96:0.0237936348,(Pasteurella_multocida:0.0651886184,Haemophilus_influenzae:0.0149019097)100:0.1377125957)90:0.0198366913)76:0.0112703769);
上面的 Newick 格式被用于重建如下所示的树。
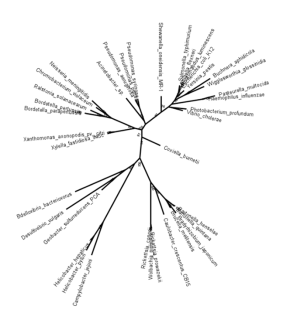
无根加性树的 Newick 格式(节点上有 Bootstrap 值)。注意分支长度精确到 10 位小数。来源:[CC BY-NC 4.0] 使用 MEGA 创建 [@mega_2021]
建树的主要方法¶
基于特征¶
建树是关于寻找分支(clades)和重建一组个体之间的系统发育关系。这些个体可以代表基因、物种或更高分类单元,但它们绝不能是类别(或平均值),因为特征和状态是对个体的观测。基于特征的方法(Character-based methods),例如最大简约法(MP)、最大似然法(Maximum likelihood, ML)和贝叶斯推断(Bayesian inference, BI),每次考虑一个特征(即 MSA 位置或列),同时比较 MSA 中的所有序列,以计算每个特征的得分。任务是找到在所有特征上具有最佳总体得分的树。这个得分也称为最优性准则(optimality criterion),是衡量数据(MSA 中的特征)与特定候选树拟合程度的指标。然后对另一棵树重复此过程,再对另一棵树重复——拟合越好,树越好。
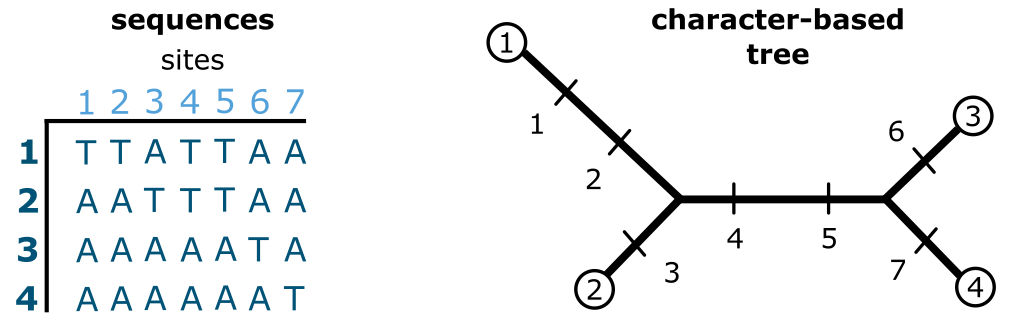
基于特征的建树。一个包含 7 个特征的 DNA 序列数据集(MSA),在 4 个末端单元(序列)上观察,使用基于特征的方法(此例为简约法)进行分析。来源:CC BY-NC 4.0 [@own_3_2024]
在上图中,给出了一个 4 个末端单元 7 个位点的 DNA 序列 MSA,以及一个起始树,每个特征都被优化到该树上。在此例中树的结构是 ((1,2)(3,4)),但也可以是 4 个末端单元的任何其他树,关键是它是一棵起始树。特征 1 和 2 是自衍征,即只在一个末端单元中出现(此例中为末端单元 1)。这意味着这两个特征与特征 3、6 和 7 一样,不贡献于(简约)树的结构,只延长其末端分支。然而,特征 4 和 5 是共同衍征,决定了这棵树的内部结构。在所有特征上,总树长为 7 步,如果使用例如 ((1,4)(2,3)) 结构(或拓扑)的树,结果可能完全不同。因此可能存在多棵同等简约的树,这就引出了下一个问题:搜索树空间。
树空间与启发式搜索方法¶
可能的二叉树(Bifurcating)数量随包含的分类单元(MSA 中的末端单元或序列)数量增加而天文数字般增长,无法通过解析方法计算(见框 3.3)。例如,10 个和 30 个序列的无根二叉树总数分别为 \(2,027,025\) 和 \(4.95 \times 10^{38}\)。实际上,很快就会变得不可能比较所有可能的树并找到精确的最优解。为克服此问题,生成随机树作为树空间中偏远且不同位置的搜索起点。不同类型的分支交换和局部重排可以用来改善树得分,然后选择得分最高的树(可能有多个)。这种树搜索方法称为启发式(heuristic,而非精确的),产生尽可能好的估计,但不保证是最优解。答案代表估计值,是否找到"最优树"仍是一个开放问题。
框 3.3:给定一组末端单元(n),可能有多少棵二叉树?
这个数字随 n 的增加非常快地增长。注意:无根("无序")树的数量遵循有根树的趋势。
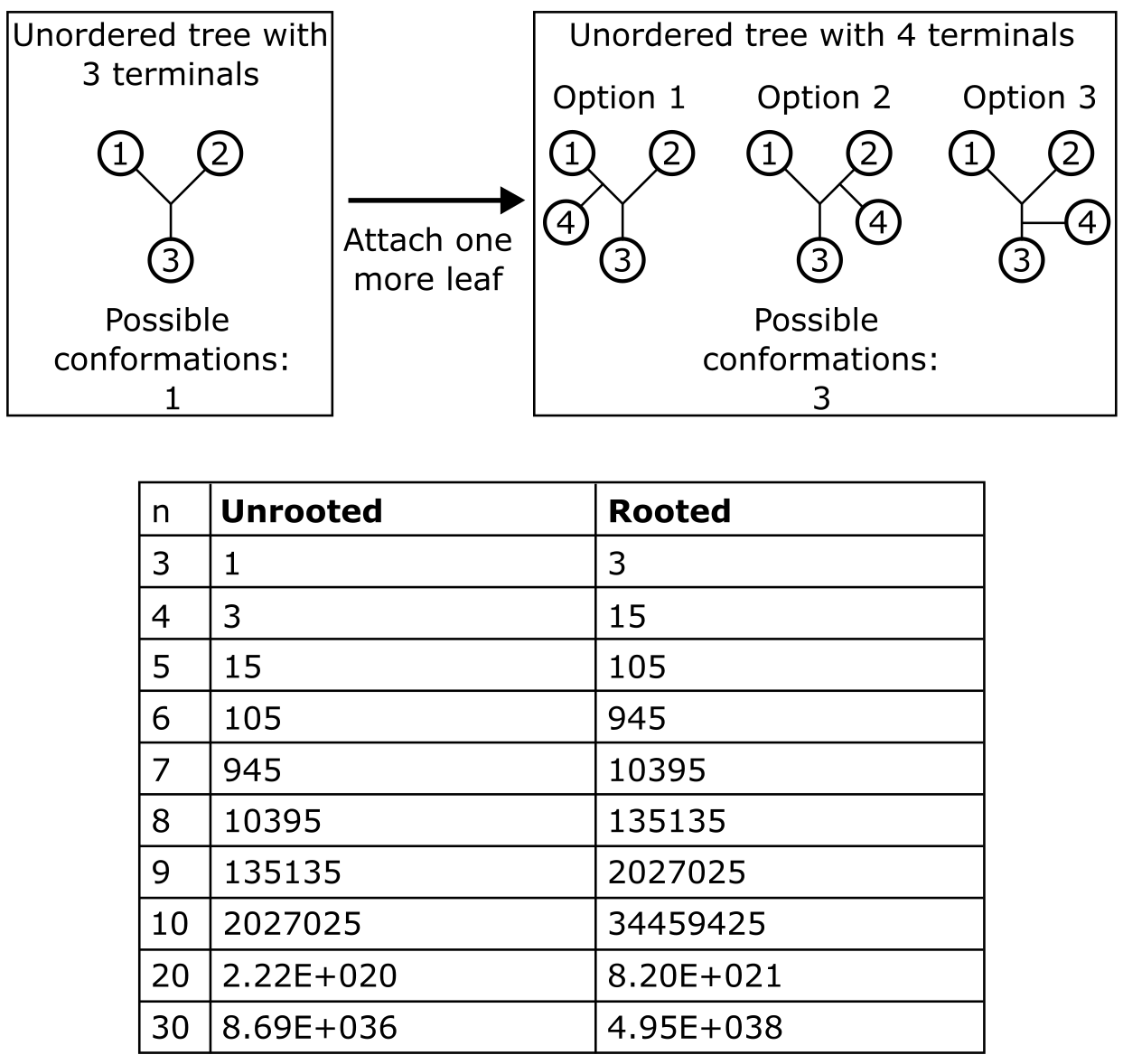
评估如此大量的树属于"NP 完全"问题类别,即使有无限资源也无法在有生之年解决。来源:CC BY-NC 4.0 [@own_3_2024]
这些基于特征的建树方法(与下面基于距离的方法相对)的优势在于,树直接由序列特征构建,能够详细分析哪些特征在树的哪些位置有贡献,或重建祖先特征(以及序列)可能是什么样的。这是基于特征的建树方法的一个强大特性,近年来已成为主导方法。
一致树¶
基于特征的建树方法通常不会只产生一棵最优树,而是一组在所应用的最优性准则下得分最优的树。在这种情况下,需要计算一致树(Consensus tree)以高效传达分析结果。下图展示了三棵树及其严格一致树和 50% 多数规则一致树。树之间的全同性意味着每棵树中都可以找到相同的节点(因此是分支)。可能存在差异,但这些差异不与其他树的拓扑结构矛盾。
树 1、2 和 3 是不一致的(即它们包含与其他树中分支矛盾的分支),因此应用正确的一致方法以可视化树之间的差异很重要。严格一致树(strict)要求只可视化相同的树拓扑结构。由于情况并非如此(AB,C 存在于树 1 和树 3 中,但不在树 2 中;'AB,C' 意味着存在一个 AB 分支,C 是其姐妹),严格一致树中该部分塌缩为三分支(A,B,C)。同样,D 和 E 仅在树 3 中单系,因此该部分在"深层"三分支(D,E,其余部分)中塌缩。严格一致树因此通常被认为过于严格,会丢失信息。对于最常用的 50% 多数规则一致树(50% majority-rule),基于每个节点在整组树中的出现频率,量化了一组树之间的(不)一致性程度,并应用多数规则阈值。因此,分支 AB 出现在树 1 和树 3 中,在 50% 多数规则一致树中的群频率为 ⅔ 或 67%。分支 ABC 存在于所有树中,得到 100%。分支 ABCD 存在于树 1 和树 2 中,得到 67%。DE 仅出现一次,得到 33%,低于 50% 的多数,因此不出现在 50% 多数规则一致树中。
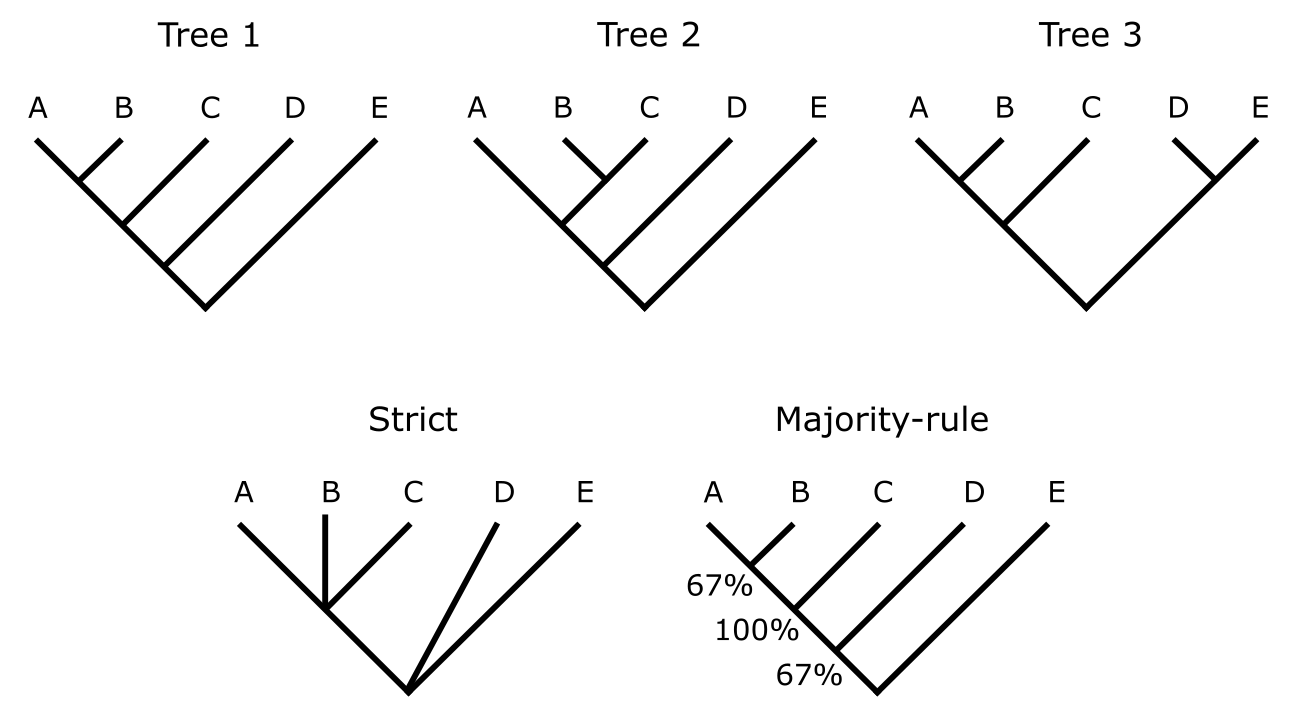
一致树。上方展示了三棵原始树,下方展示了它们的严格一致树和 50% 多数规则一致树。来源:CC BY-NC 4.0 [@own_3_2024]
简约分析¶
基于特征的建树最简单的方法是简约分析(parsimony analysis),逐个特征地计算每个特征在候选树上的拟合度(步数)。有些特征可能只变化了一次但在多个序列中同时出现(共同衍征),而另一些可能独立变化了多次(称为同塑性,homoplasies)。有些特征可能只在一个序列中变化(自衍征,autapomorphy)。当 MSA 中的所有特征都被评估后,通过将所有特征的变化相加计算数据与候选树的总体拟合得分。然后假设另一棵候选树并再次执行该过程。越来越多的树以这种方式进行比较,直到剩下单个最优或一组同等简约的重建结果。鉴于即使中等数量末端单元的树空间也极其庞大(见框 3.3),此过程可能需要一些时间完成。通常在超过 15 个末端单元的情况下,只应用启发式搜索方法(见树空间与启发式搜索方法)。
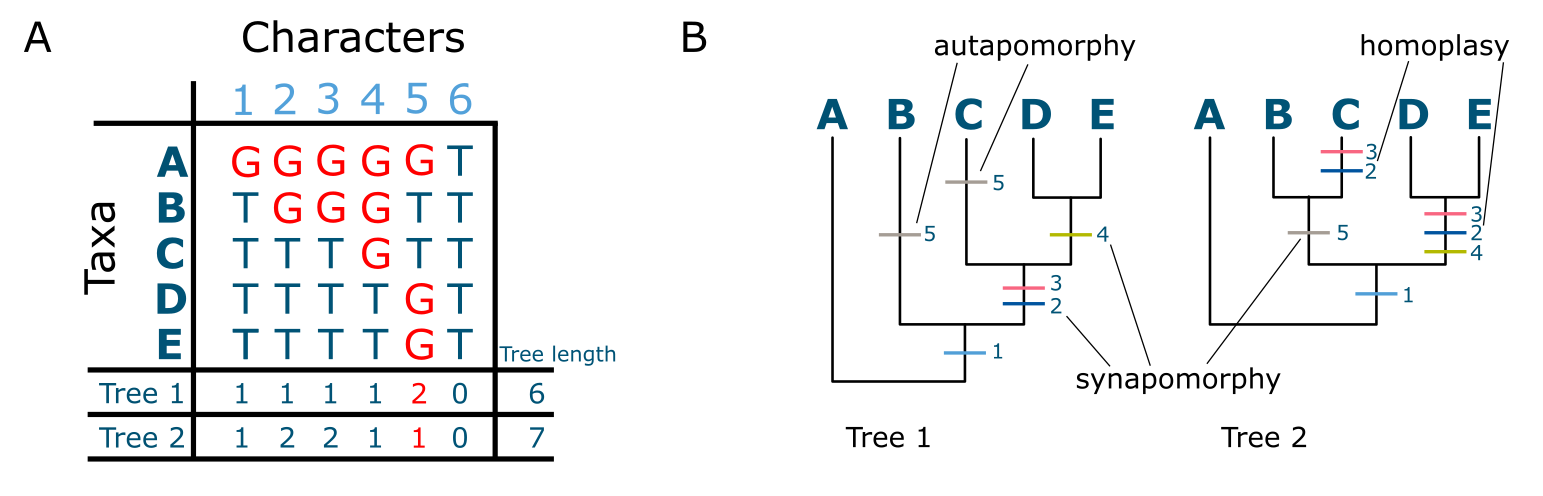
简约分析(数据与树与上图相同)。MSA 中所有特征的状态变化("步")标在树上,示例共同衍征和自衍征已标出。注意特征 6 是不变的,因此不对任何树有贡献。另注意 MSA 中每次替换在树上产生额外一步。来源:CC BY-NC 4.0 [@own_3_2024]
每个特征变化都可以被视为一个特设假设(ad hoc assumption),每个都与其 I 类错误(即假阳性的概率)相关联。这将是在树上推断出特征变化(替换),但实际上该位置没有发生任何变化。最小化变化数量的树也最小化了特设假设的数量,因此也最小化了 I 类错误。

奥卡姆的威廉,"简约之父",14 世纪。来源:CC BY-SA 4.0 [@ockham_2022]
这就是简约准则,已在 14 世纪由方济各会修士奥卡姆的威廉(William of Ockham)描述,并被称为"奥卡姆剃刀"(Occam's razor)。换言之:当面对关于同一预测的竞争假说时,应选择假设最少的解决方案。
自然界是否简约?这是一个常见的问题,可能受到奥卡姆剃刀本身可能是一个假设的观点的启发,但需要记住的是,简约准则仅应用于在假说(即树)之间做出选择,并且不对自然界和进化做任何假设!最大简约方法包含在本章实践所使用的软件 MEGA11 中 [@mega_2021]。
还有两种重要的基于特征的建树方法:最大似然(ML)分析和贝叶斯推断(BI)。两者与简约分析的不同之处在于,它们不仅计算差异(如简约分析),而是基于显式的特征进化模型并在概率框架下运作。ML 将在下文的最大似然建树一节中讨论;BI 超出本课程范围,因此不在此处理。
基于距离¶
建树的另一个主要方法是聚类(clustering),是基于距离(distance)的,在多个应用中广泛使用,例如将 BLAST 搜索可视化为邻接树(Neighbor Joining trees)。在基于距离的方法(Distance-based methods)中,不是一次比较一个特征,而是对所有可能序列对进行整个序列的成对比较(即同时比较所有特征),通常产生一个三角形的全对全距离矩阵。成对比较产生成对距离,可以是超度量距离或欧几里得距离(见框 3.4)。请注意距离(\(D\))与相似性(\(S\))之间的关系:
在不同研究中,\(D\) 或 \(S\) 可用于比较。使用的是哪一个通常可以从结果成对距离矩阵的对角线推断,其中每个序列与自身比较。距离矩阵对角线为全 0,相似性矩阵为全 1。
在上图中,通过对每个可能序列对中差异数的计数,从与上图中相同 MSA 计算成对距离。这产生一个三角形成对距离矩阵,其值(距离)随后用于构建距离树,例如使用邻接法(Neighbor Joining)。MSA 此后不再用于分析。在此例中,距离值完美拟合结果距离树。
注意上图中的树和上图中树的拓扑结构相同,但简约树包含更多信息:除了分支模式和分支长度外,它还包含哪个特征在树的哪个位置发生了变化的信息。
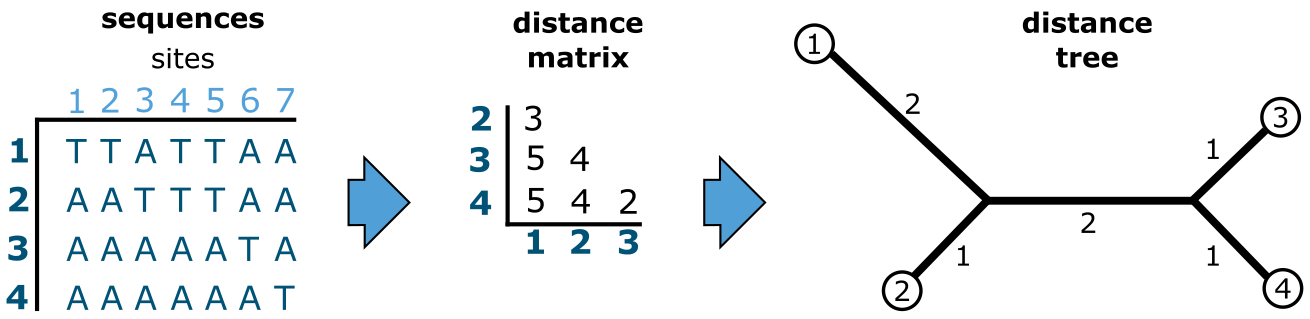
基于距离的建树。上图中的 7 个特征 4 个末端单元(序列)的相同数据集(MSA),使用基于距离的方法进行分析。来源:CC BY-NC 4.0 [@own_3_2024]
如果序列以时钟式方式积累替换(即像放射性衰变),结果成对距离甚至可能是超度量的(见框 3.4)。这意味着三角形成对距离矩阵中的距离与在结果距离树上测量的距离相同。然而,这种干净的数据几乎从未被找到,在树上测量的距离可能与成对矩阵中的观测距离不同。下图展示了两棵树:超度量树(A)和加性树(B),后者包含不等长的姐妹分支(到 a 和 b)。在加性距离矩阵(矩阵 B)中,由于到 a 和 b 的长度差异,最相似的序列(即 b 和 c)实际上可能不是最近缘的(即 a 和 b)。
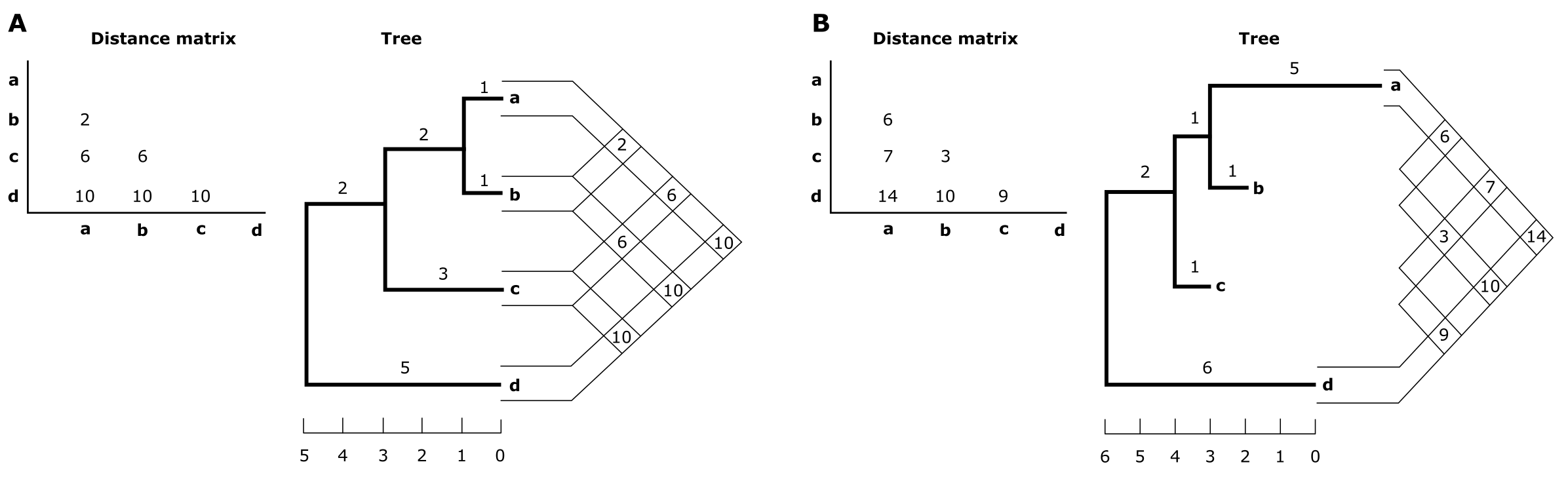
(A) 四个序列 a-d 之间的超度量距离矩阵及对应的超度量树。(B) 四个序列 a-d 之间的加性距离矩阵及对应的加性树。距离矩阵中的值对应于树上两个序列之间路径的分支长度之和,因此此数据是度量的。注意在 B 中,b 和 c 最相似,但不是最近缘的(a 和 b 才是最近缘的)。来源:CC BY-NC 4.0 [@own_3_2024]
一旦这些成对距离被计算,MSA 就不再使用,树直接从距离构建。与基于特征的树不同,在基于距离(聚类)的树中,无法评估哪个特征在树的哪个位置有贡献,因为所有个体特征都已合并为一个总体成对距离值。此外,不变特征(MSA 中不包含变异的位置)确实对成对距离值有贡献。这是与基于特征方法的主要差异,后者只有可变特征对树有贡献。
聚类方法的优点是速度快,不需要大量计算资源(不存在树空间或 NP 完全性问题,如上所述的基于特征方法(见框 3.3))。聚类方法将个体分配到簇中,使得同一簇中的个体比其他簇中的个体更相似。没有显式得分或最优性准则,只有所有序列间总距离的最小化。聚类通常只产生一棵树,不显示替代的"同等好"的树;这是由于聚类算法设计为产生单棵树。
框 3.4:距离度量及其性质
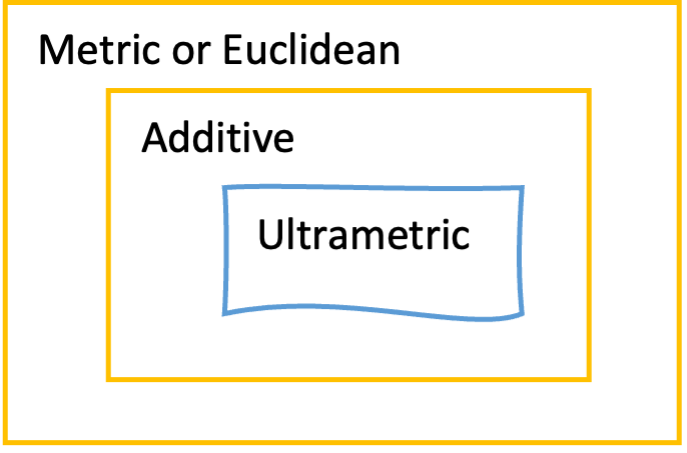
来源:CC BY-NC 4.0 [@own_3_2024]
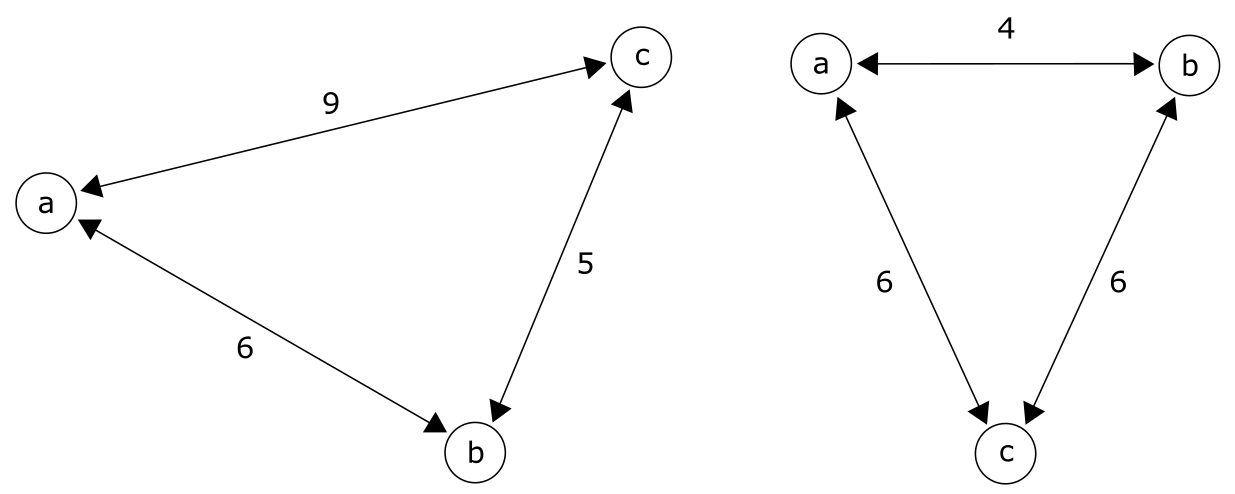
来源:CC BY-NC 4.0 [@own_3_2024]
欧几里得距离或度量距离(metric distance)要求观测距离是非负的、对称的、互不相同的,并满足三角不等式(Triangle inequality):任意一对序列 a 和 b 之间的距离不能超过它们与第三个序列 c 之间距离之和。
超度量距离(ultrametric distance)以超度量不等式(Ultrametric inequality)为特征:在比较三个序列时,两个最大距离相等。超度量距离具有吸引人的特性,它们以时钟式进化,因此最相似的序列也将是最近缘的。事实上,超度量树完美描述了距离矩阵中所示的观测距离。
邻接法¶
可能最常用的距离建树方法是邻接法(Neighbor Joining, NJ),它快速有效,尤其适用于大型 MSA(包含数百个序列)。NJ 建树从一个完全未解析的树开始,包含 MSA 中的所有序列,通过对所有成对距离求和计算总树长(或总体起始距离)。然后选择一对序列合并以开始一个小簇("邻居"),并更新总树长,用合并的邻居替换原来的两个末端单元。重复此步骤直到所有序列和序列对都被合并,同时最小化它们之间的总体距离(树长)。
邻接法产生无根树,因此如果需要根化,应使用外群根化。没有分子钟假设,允许邻居(姐妹)之间的分支长度差异被重建。NJ 在 MEGA11 中实现 [@mega_2021],并在实践中使用。
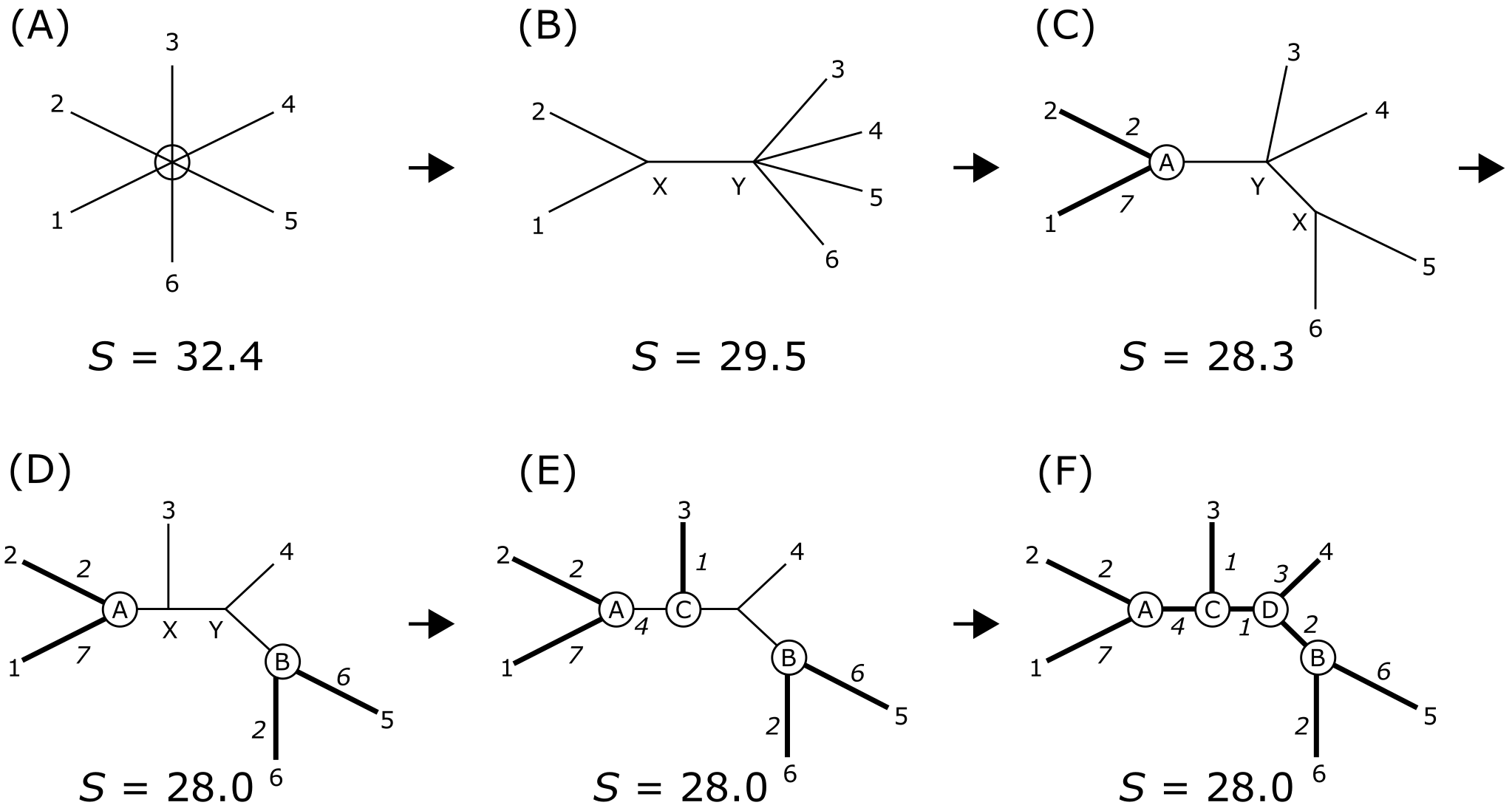
邻接法。计算过程的图示。树长 S 是所有分支长度之和,通过从星树(A)开始迭代合并邻居来最小化(F)。来源:CC BY-NC 4.0 [@own_3_2024]
NJ 非常流行,因为它可以在很短时间内生成包含数百个末端单元的树。这使其成为快速评估数据集(MSA)中(系统发育)结构的理想工具,而无需探索广阔的树空间(如基于特征的方法)。需要注意的是,NJ 是一种聚类方法,即基于总体相似性对序列进行分组,而非基于共同祖先或共同衍征(Synapomorphy)。因此,对于系统发育研究,首选基于特征的分析,NJ 分析可以作为补充,检查两者之间可能的不一致性。如果发现不一致,可能意味着数据(MSA 中积累的共同衍征)在树的该部分不是度量的,可能需要额外的分析方法(如系统发育网络重建),这超出了本课程的范围。
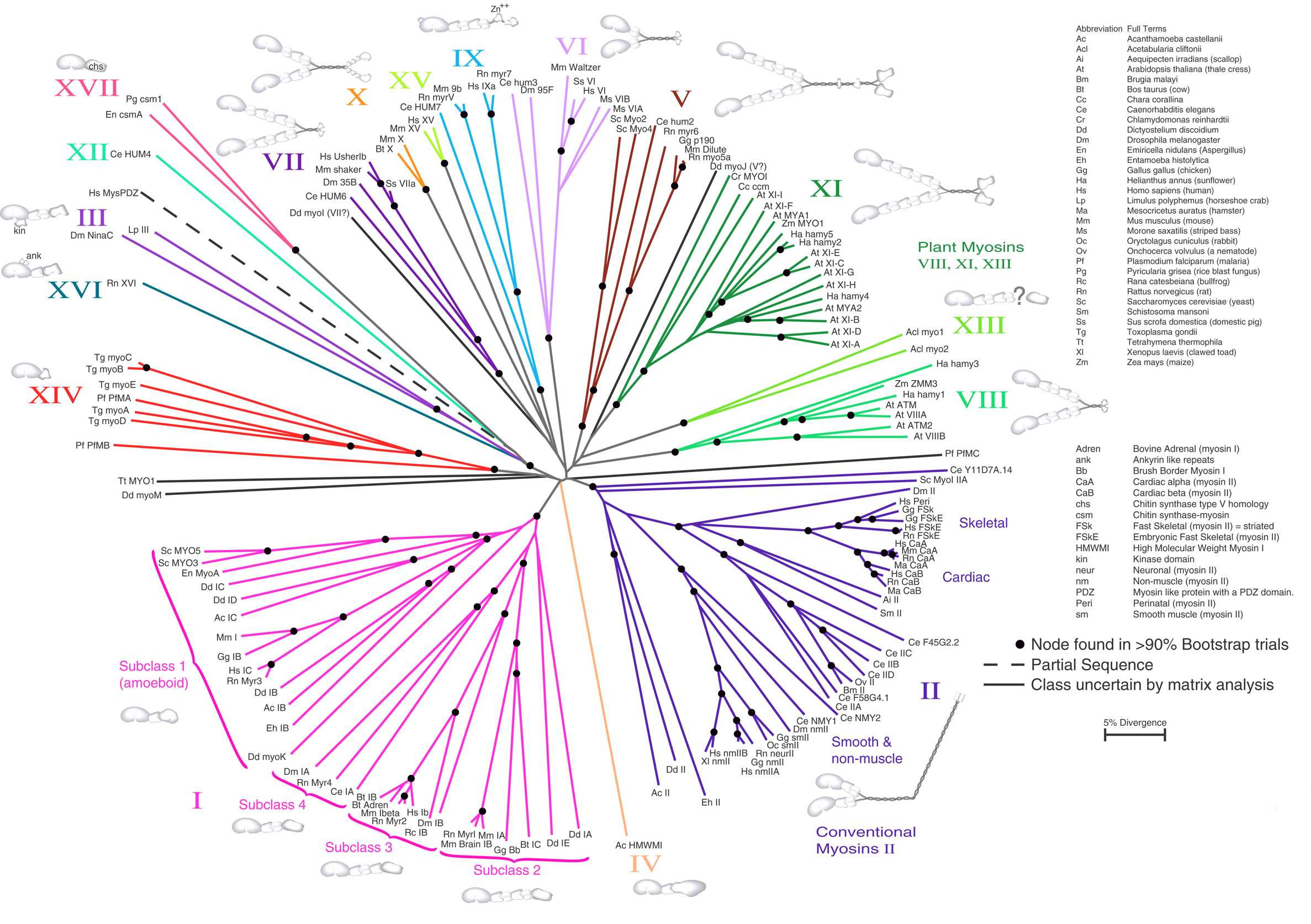
邻接法。基于肌球蛋白氨基酸序列的无根 NJ 树。标尺表示 5% 的序列分歧度。来源:CC BY 1.0 [@neighbor_joining_aa_2000]
估计序列分歧度¶
在系统发育中,当长末端分支与短内部分支混合时,使用核苷酸和简约分析的系统发育重建通常是有问题的。原因在于,当使用 DNA MSA 时,在长分支(即高度分歧的序列)上,4 个核苷酸中任何一个在两条分支上随机出现的概率实际上相当高,可能导致假共同衍征(false synapomorphies)。当积累了一些之后,可能导致错误的分支。这种所谓的长枝吸引(Long Branch Attraction, LBA)假象相当常见,尤其是在涉及孤立的老谱系(如被子植物中的 Amborella 或 Nymphaea)时,但也可能出现在基因树中。已证明 LBA 可以通过建模分支长度(而非像简约分析那样仅将差异计数作为分支长度,每次替换在 MSA 中导致树长额外一步)来在一定程度上缓解。然而,为了准确估计系统发育树中的分支长度,我们需要准确的序列分歧度估计。
同源序列之间的进化分歧度(或距离)反映在它们自 MRCA 分裂以来的替换中。直观地说,在比较两个序列时,可以直接取差异位点的比例作为序列分歧度,例如,对于 1000 个位置的序列,有 10 个差异将产生 0.01 或 1% 的差异。然而,这种所谓的 p 差异(p-difference)不一定考虑了两条序列分歧期间历史上发生的所有替换,其中可能包括回返原始状态的变化。估计"真实"序列分歧度意味着我们需要找到确实发生但在你的 MSA 中不可见的替换。可变位点实际上可以在进化过程中持续变化,导致同一位置发生多次替换,这可能导致变化饱和。这样,多次替换可能被忽略,仅用 p 差异会低估实际序列分歧度。
替换模型¶
替换模型都基于下面给出的 Jukes-Cantor(JC)公式,对未观测事件进行分歧度估计的校正。JC 公式基于计算特定位点发生替换的概率加上该位点不变化为其他三个核苷酸中任意一个的概率。公式中,\(p\) 代表观测到的差异比例(即 p 差异),\(d\) 代表校正后的分歧度度量。当所有位点都不同(即 \(p = 1\))时,\(d\) 在极限下达到 0.75,即校正后的 \(d\) 不能超过 75%。
下图展示了 JC 公式如何在替换模型(JC 模型)中应用。有一个矩阵定义了 4 个核苷酸碱基之间六种可能的替换类型,即 T↔C、A↔G、A↔T、T↔G、C↔G 和 C↔A。在此情况下,所有六种替换类型的相对速率被假设为相等,并用一个共享参数表示,称为'a'。该模型的另一个假设是 MSA 中的碱基组成相等,假设每个碱基在每个位置的出现概率为 25%。JC 模型被认为是一个相当简单的单参数模型。
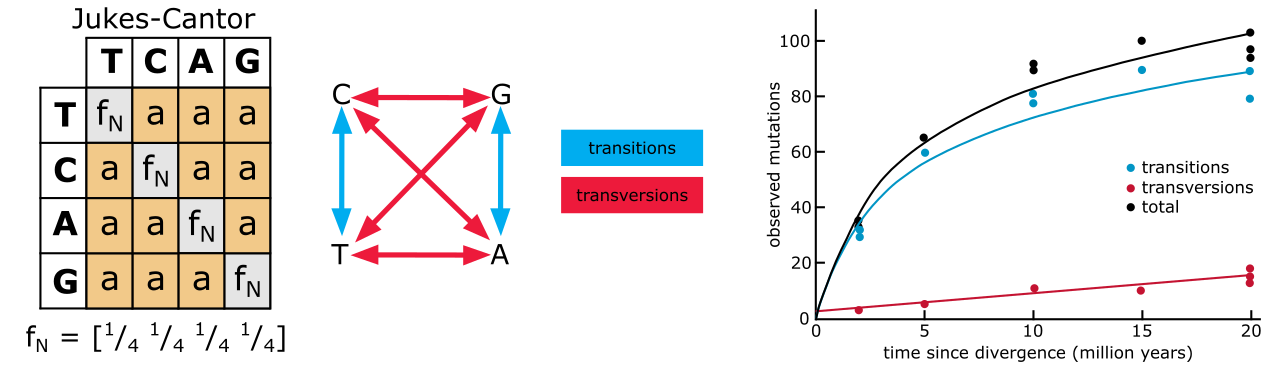
Jukes Cantor 模型(左),转换(蓝色)和颠换(红色)以及它们在进化时间中的不同积累(右)。来源:CC BY-NC 4.0 [@own_3_2024]
上面列出的前两种替换类型是转换(Transition,嘧啶 T 和 C 之间、嘌呤 A 和 G 之间的替换),而其余四种发生在嘌呤和嘧啶之间,称为颠换(Transversion)。转换速率(ti)与颠换速率(tv)有不同的动态,因此替换的积累也不同。在 Kimura 2 参数(K2P)模型中,通过添加额外参数 b 来解决此问题。参数 a 估计 ti(\(P\)),参数 \(b\) 估计 tv(\(Q\));在 Kimura 2 参数公式中,\(P\) 和 \(Q\) 分别是 ti 和 tv 的比例:
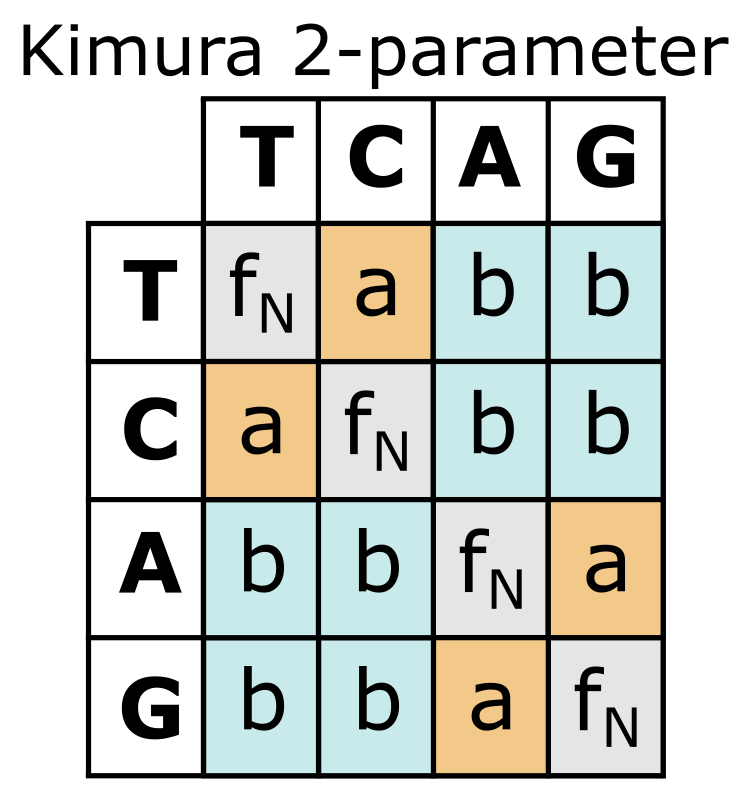
Kimura 2 参数替换模型,转换以橙色标出(参数 a),颠换以蓝色标出(参数 b)。注意此模型中碱基频率 fN 被视为相等。来源:CC BY-NC 4.0 [@own_3_2024]
除了 JC 和 K2P 模型外,还存在考虑 DNA 序列进化不同方面的其他模型,例如所有六种替换类型之间的差异(广时间可逆模型或 GTR)、序列碱基组成或核苷酸频率(Felsenstein81 或 F81)。另一个方面是 MSA 中位点变化速率的分布:有多少快速变化和慢速变化的位点,以及它们如何分布。通过与伽马 Γ 分布比较来实现(未显示)。最复杂的模型包含许多参数,由 DNA 序列进化所有这些方面的组合构成。有高达 220 种不同模型可供选择。需要认识到的是这些模型是可逆的,因此只允许重建无根树。一旦确定,可以使用外群根化来根化它们。
对于氨基酸序列比较,不是从数据中估计参数值,氨基酸替换模型基于(预定义的)替换代价矩阵(见第 2 章),这些矩阵基于在超过 30,000 个蛋白质序列中观察到的氨基酸替换(例如 JTT、Blosum、Dayhoff、LG 和 WAG 矩阵)。
最大似然建树¶
对于基于特征的方法,这些替换模型基于概率,使我们能够计算数据支持特定树和模型的似然(likelihood)。这个似然是一个描述数据、树和模型拟合程度的得分:
用文字描述:似然 \(L\) 是在给定假说 \(H\)(包括替换模型和所选树)的情况下获得数据 \(D\) 的概率。因此也可以说:
显然,为数据集(MSA)选择最佳拟合模型很重要。模型选择通过使用一系列不同模型和同一棵树计算 MSA 的似然来进行;最佳似然得分意味着最佳拟合模型。
随后考虑候选树,并在给定该模型和特定树的情况下计算观测数据(MSA)的似然 LD。然后考虑另一棵树,同时保持选择相同的最佳拟合模型,再次估计其参数值。再次计算观测数据(MSA)的似然 LD,这次似然可能更好。评估更多的树,考虑更多的模型参数值,持续追踪 LD 直到无法获得进一步增加。这通常通过上面概述的启发式树搜索方法来实现,并取决于由 MSA 中序列数量决定的树空间。最终结果是最大似然估计(MLE):使数据似然最大化的树和模型参数值的组合。这棵树(可能不是精确的最佳 MLE——毕竟使用了启发式方法)通常被称为 ML 树。
基于 MLE 的系统发育树重建在过去十年中已成为主导的建树方法。它是一种高效的方法,可以考虑 MSA 中序列之间替换速率和模式的差异。这意味着非时钟式或偏向(非随机)的替换积累将被建模,这将最小化推断 ML 树拓扑结构时可能的假象,例如 LBA(见上文)。
系统发育重建的 MLE 流程在软件包 IQ-TREE 中实现 [@iqtree_2020],包括 I) 模型测试,II) ML 树搜索,和 III) 核苷酸和氨基酸序列的 Bootstrap 分析。IQ-TREE 将在本章实践中演示和使用。
最后,使用核苷酸替换模型估计进化分歧度也可以应用于距离方法。在那里,替换模型被应用于计算"校正的"成对序列距离,然后用于产生邻接树。
模型测试、ML 树搜索、Bootstrap 分析¶
在获得一棵带分支长度的 ML 树后,仍然没有关于 ML 树中节点在数据(MSA)支持方面可能如何不同的信息。因此进行 Bootstrap 分析,基于从 MSA 中抽取的伪重复数据集重复 MLE 过程多次。在为每个伪重复数据集获得 ML 树后,计算 50% 多数规则一致树以查看群频率(每个节点在重复中出现的比例)。这些频率也称为 Bootstrap 值(bootstrap value)。其思想是节点的共同衍征越多,其 Bootstrap 值越高。
不幸的是,特征支持与 Bootstrap 值之间没有简单的线性关系。通常,Bootstrap 值 < 90% 被认为对该节点的支持度差,< 50%(甚至 < 60%)被视为"无支持"。含有一个共同衍征的 MSA 通常获得 62% 的 Bootstrap 值,意味着这类节点可能应该被忽略,但这取决于分析师的经验和解读。下图展示了当弱支持的节点被塌缩时 Bootstrap 一致树的变化:树拓扑结构解析度降低,但保留的部分是强壮的。这确实是一个权衡:可视化大量好看但支持度差的解析度,还是只关注强节点。通常,我们希望两者都看到。
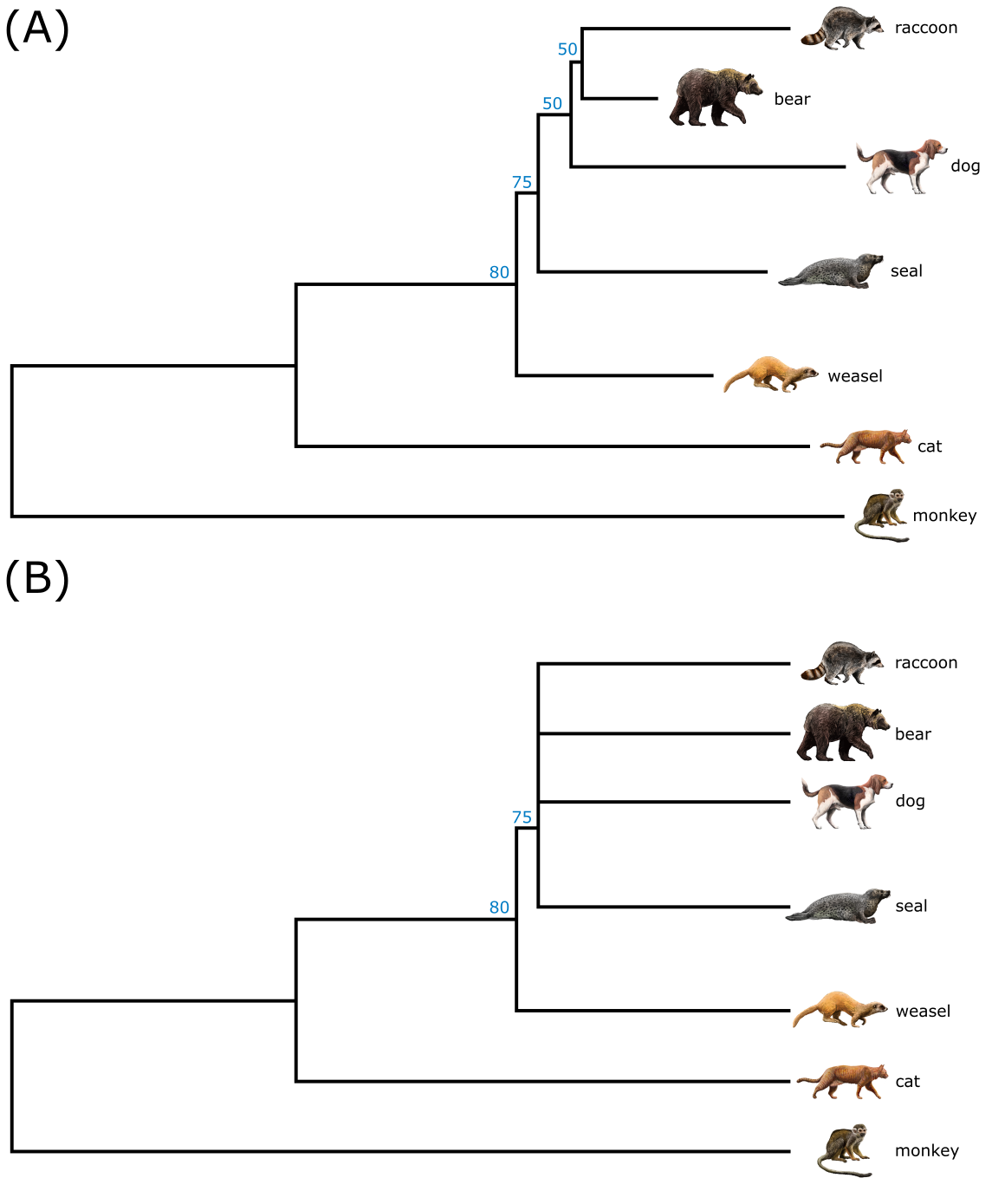
Bootstrap 分析。A) 一棵最大似然树,节点上标出了 Bootstrap 值。注意并非所有节点都显示 Bootstrap 值,可能是因为 < 50% 的值被忽略了。B) 同一分析,但这次所有 Bootstrap 值 < 50% 的节点被塌缩。注意塌缩弱节点后产生的包含四条谱系的多分支;以及塌缩树中从加性树到分支图样式的变化。来源:CC BY-NC 4.0 [@own_3_2024]
建树方法总结¶
总结来说,建树方法可以根据使用的数据类型分类:特征(或"位点")对比成对距离(聚类分析)。使用特征建树需要应用最优性准则以在多个(有时是巨大的)候选树中选择。聚类通常产生一棵树,最优性准则方法产生多棵,此时需要计算一致树。
另一个有用的分类标准是是否应用了显式的特征进化模型,对比仅计数(简约)变化。在本课程中我们涵盖了最大似然和简约法,以及使用校正距离的邻接法,即在成对序列比较中应用特征模型。贝叶斯推断(计算节点概率并同时评估不同模型)超出本课程范围。(如果你感兴趣,它们在 BIS-40306 比较生物学与系统学中涵盖)。
| 距离(成对) | 位点(特征) | |
|---|---|---|
| 方法 | ||
| 显式特征进化模型 | 邻接法 邻接网络 | 最大似然 贝叶斯推断 |
| 无特征进化模型 | p 差异 | 简约法("计数") |
| 最优性准则 | 否,聚类 | 是,树长或似然 |
实践作业¶
本指南包含帮助你处理第 3 章学习材料的问题和练习。你有两个上午的时间完成这些练习。在单次会话中,你应该完成大约一半的指南,即作业 I-IV。注意作业 VI 是可选的。
这些实践练习为你提供了第 6 周项目的最佳准备,工具及其使用也是考试内容的一部分。因此,请确保你现在培养实践技能,以便在项目期间应用它们,并在考试中展示你的观察和解读技能。
注意,答案将在实践课后发布!
在 MEGA11 中构建基于氨基酸的 PLT1 树并在 iTOL 中可视化,30 分钟
首先,在这个作业中,你将表达一组 10 个 PLT1 氨基酸序列之间的关系,你在上一章中已经为这些序列生成了多重序列比对(MSA)。你将通过使用 Molecular Evolutionary Genetics Analysis(MEGA 11)构建简约树来完成此任务。然后使用树图形软件 FigTree 和 iTOL 可视化你的树,iTOL 是一个非常有用的平台,用于存储、管理树并制作漂亮的树图。
首先,启动 MEGA 11,打开并查看可用的标准 MEGA 11 仪表板。白色开放区域作为画布,每个分析将显示为固定便签。除"PHYLOGENY"外还有许多有趣的工具,其中一些你今天将使用。
使用 File > Open,打开你上一章的 PLT1 MSA;这个文件应该是一个 FASTA 文件,文件名扩展名为 .fasta。当在 Analyze 和 Align 之间选择时,选择 Analyze(因为序列已对齐)。然后选择正确的数据类型。
- 通过选择
Data > Explore Active Data(或点击TA按钮)查看你的比对,这将进入 DataExplorer。 - 使用设置
Display > Color Cells你可以清楚地看到氨基酸如何对齐,以及高保守区域如何与高变异区域交替。 - 点击
TA按钮可以高亮每个比对位置上非相同的氨基酸,这在 100+ 序列的大型比对中特别有用。
现在关闭 Data Explorer,返回 MEGA 仪表板,点击 Phylogeny > Construct/Test Maximum Parsimony Tree(s)。保持默认设置,包括"no test of phylogeny"(Bootstrap)和"MP search Method"为 SPR 分支交换。
- 确认并使用默认设置计算树。
几秒钟后你将看到你的树,它是分支图样式(没有分支长度信息,仅拓扑结构)。为了查看分支长度,选择"Layout"(左上窗口),然后 Toggle scaling of the Tree,这将计算这棵树的分支长度;这应该在 2 分钟内完成。
选择"Branch lengths",你可以看到树上分支上的平均氨基酸替换数(每个位置多个可能优化的平均值)。
MEGA 的一个好处是点击 Caption(左下角)你将获得此分析中使用的所有设置以及结果的完整信息;例如,有多少个最简约重建结果(此例中只有一个),起始树如何计算以及 MSA 中有多少位点。
- 现在保存你的树:选择
File > Export Current Tree(Newick),勾选"Branch length"框,点击"Ok"。Newick 格式将显示在文本文件编辑器中,选择File > Save as,给它一个合理的名称,将 .nwk 扩展名替换为 .tre。 - 你可以重复最大简约分析,但这次在"Phylogeny Test"处选择
Bootstrap method。这将执行 500 次重复的 Bootstrap 分析。 - 完成后,你将在树窗口上方看到两个标签:'Original Tree' 显示初始简约结果,Bootstrap 值叠加在(一些)节点上。另一个标签名为'Bootstrap consensus tree',显示实际的 Bootstrap 分析。
- 使用 Layout >
Toggle scaling of the tree查看平均分支长度。 - 如上保存你的 Bootstrap 树,这次勾选
Bootstrap Values框。
我们在课堂上看到,< 50% 的 Bootstrap 值实际上相当无信息量,因为单个共同衍征就可能得到 62% 的结果。因此,查看我们树的"浓缩"版本会比较好。
- 确保你在 Original tree 标签中,然后点击
Compute > Compute condensed tree按钮(左上角)。你将得到"Tree Cutoff options",在 % cutoff 框中输入 62。看你的树发生了什么变化?
问题:
- 为什么 Bootstrap 一致树的拓扑结构与最简约树不同?
- 你的浓缩树中的多分支代表什么?
可视化你的树是系统发育分析的关键步骤,因为树中包含的(层级)信息本质上是图形化的。系统发育树图像的重要方面包括:
- 树结构:在矩形样式的树中最清晰,但当分类单元(末端)数量较多时,树无法放在一页上,圆形树(分支图样式)可能是更好的解决方案。
- 分支长度:表示你的分类单元或基因上发生的进化量,因此本身就有价值;同样,当末端数量较大时,圆形树(带加性分支长度)可能是更好的解决方案。
- 节点支持:通常显示 Bootstrap 值(群频率),但也可以用符号或节点下分支的按比例宽度来表示。
- 根化:外群根化的效果应清晰可视化。
FigTree\ 一个稳定且实用的树可视化工具,完全免费,是 FigTree,可以从这里获取。Brightspace 上包含一个视频,介绍如何使用 FigTree 进行带 Bootstrap 值的树的可视化,见明天的 IQ-TREE 实践。
iTOL\ iTOL 平台提供了非常有用的解决方案,可以在你的树图中容纳所有这些方面。我们将在这里了解其中一些功能。iTOL 的大多数功能是免费的,但保存结果和树视图需要订阅。
现在,前往 iTOL 并注册一个(免费)账户。在欢迎页面,到处看看,了解大树如何显示,以及额外的(外部)数据如何与树图形集成和注释。也请查看 Gallery,其中展示了用户上传的树。
前往 MyTrees,上传你刚才在 MEGA 11 中制作的两棵树。点击其中任何一个将可视化你的树。注意:可以将控制面板拖到一侧以查看整棵树。使用 previous 和 next tree 按钮(在 My Trees 旁边)在两棵树之间切换。虽然在你的 PLT1 MSA 中没有包含或选择外群,但树看起来有根,因为在 iTOL 中控制面板 Mode 下默认选择了"Rectangular"。因此,你需要通过选择"Unrooted"来显示无根树。
- 在
Branch options > Line style下增加线粗至 20 px,使树结构更清晰。 - 在
Labels下在显示和隐藏之间切换,以便仅查看树结构;使用 + 工具(屏幕右上角)放大。 - 在
Mode options > Rotation下你可以旋转你的树。 - 现在确保选择你的 Bootstrap 一致树,前往
Advanced > Branch metadata > Display > Bootstraps / metadata,选择"Display"然后"Text"。将字体大小增加到约 60px,四舍五入到 2 位小数,你可以看到你的 Bootstrap 值出现在树上。 - 前往
Advanced > Node options,为你的叶节点和内部节点都选择"Display"。选择一个符号和颜色,将符号大小增加到约 50px 或更大。 - 现在切换到你的"Original tree",使用
Advanced > Branch metadata display > Branch lengths,选择"Display"。将字体大小增加到约 60px,小数位四舍五入到 2 位;这样你可以从树上读取分支长度。 - 返回
Basic,确保"Mode"是 Circular 或 Rectangular(而非 Unrooted),然后选择Branch options > Connections between nodes are curves。 - 前往
Basic > Mode options,在分支长度处选择"Ignore"以查看你的树的分支图版本,即没有按比例的分支长度。注意:这不适用于你的 Bootstrap 一致树,因为它没有分支长度。(带分支长度的树要有趣得多!) - 点击树中任何内部节点,选择
Branches > This node > Colour以给树中的分支着色。 - 探索 iTOL 提供的其他树可视化功能;当你满意你的树时,使用
Export > Full image > Export并以 .svg 格式保存,以便在 Word 或 PowerPoint 文档中使用。
- 将末端标签切换回来(使用 Basic > Labels > Display),可以清楚看到对于此图,序列名称太长了。这最好在你开始使用的 .fasta 文件中手动编辑。
- 最后:如果你有外群信息,可以通过选择一个节点或分支(可以是内部和末端节点),然后选择
Editing > Tree structure > Re-root the tree here来在树上放置根。如果你想再次重新根化,需要先重置树(在控制面板中右下角黄色按钮)。
通过这些步骤,你已经从 MEGA 11 生成的树变为了漂亮的树图,这是一个你可以应用于本实践中生成的其他树以及当然课程后期项目中的流程。同时,使用 iTOL 存储你的树并保持对所有生成树的概览。
估计序列分歧度:探索 MSA,20 分钟
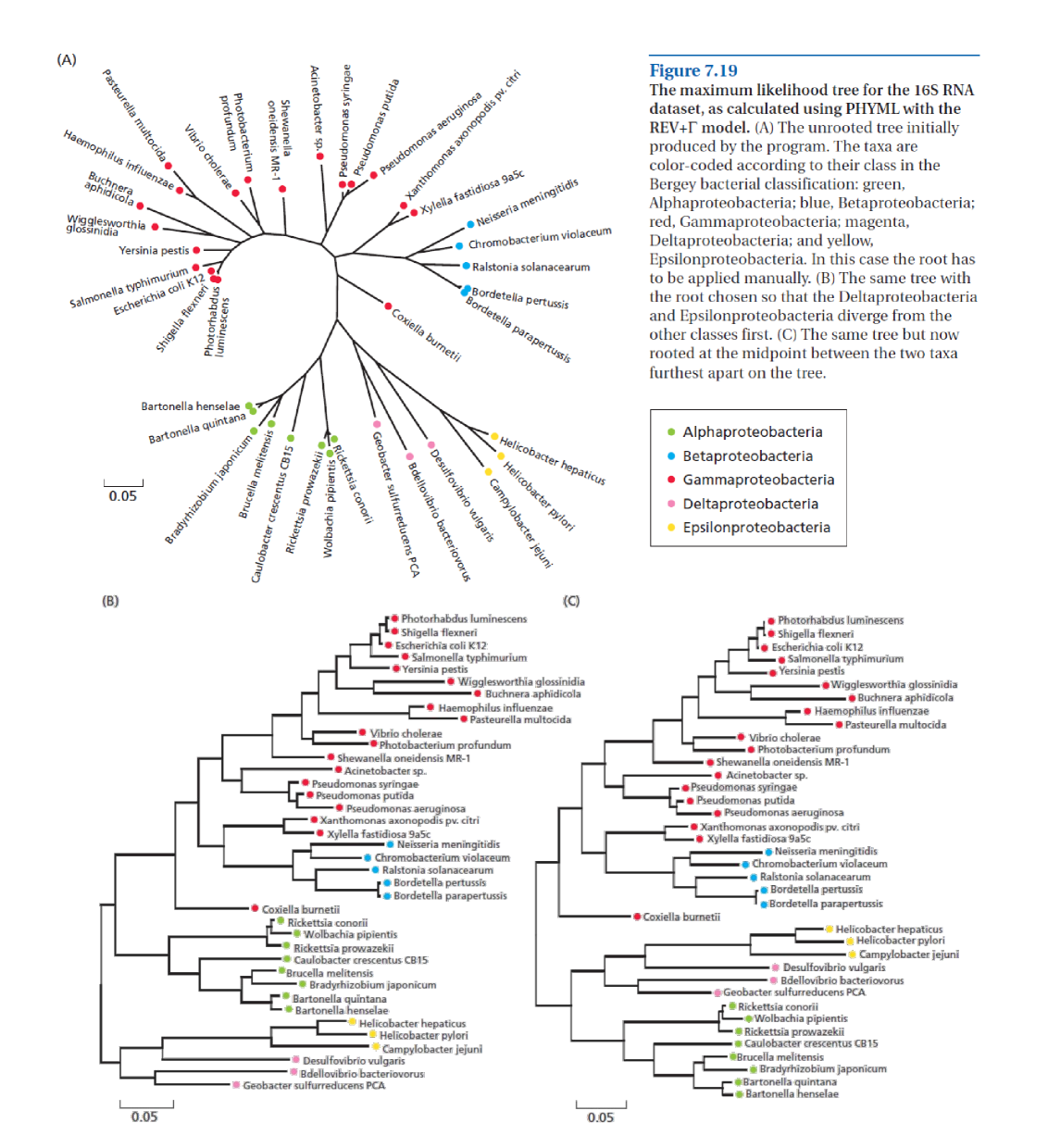
变形菌 16S rDNA 序列(见上图,源自 [@bioinformatics_2007])已被编译和比对,涵盖变形菌门(Proteobacteria)38 个细菌物种,该门通常分为五个纲:α-、β-、γ-、δ- 和 ε-变形菌(见这里了解该门的进一步背景)。此处包含的 38 个物种分布在这五个纲中,因此系统发育重建应该能够恢复五个分支,如果这种分类确实基于进化的话。在本实践中你将估计这 38 个 rDNA 序列之间的序列分歧度,然后构建系统发育关系。
首先你将探索多重序列比对(MSA)中的分歧度,以及如何对原始的比例 p 差异进行校正,以估计它们的"真实"进化分歧度。
启动 MEGA 11,打开 BrightSpace 上提供的 16S rDNA 序列比对(通过选择 Data > Open A File/Session)并选择 Analyze。然后选择合适的数据类型,记住 16S rDNA 不是蛋白编码序列。现在使用 DataExplorer(Data > Explore Active Data)探索 SSU 序列比对。使用设置 Display > Color Cells 你可以清楚地看到核苷酸如何对齐,以及高保守区域如何与高变异区域交替。(这对于后面选择正确的核苷酸替换模型来计算似然树时相关)。
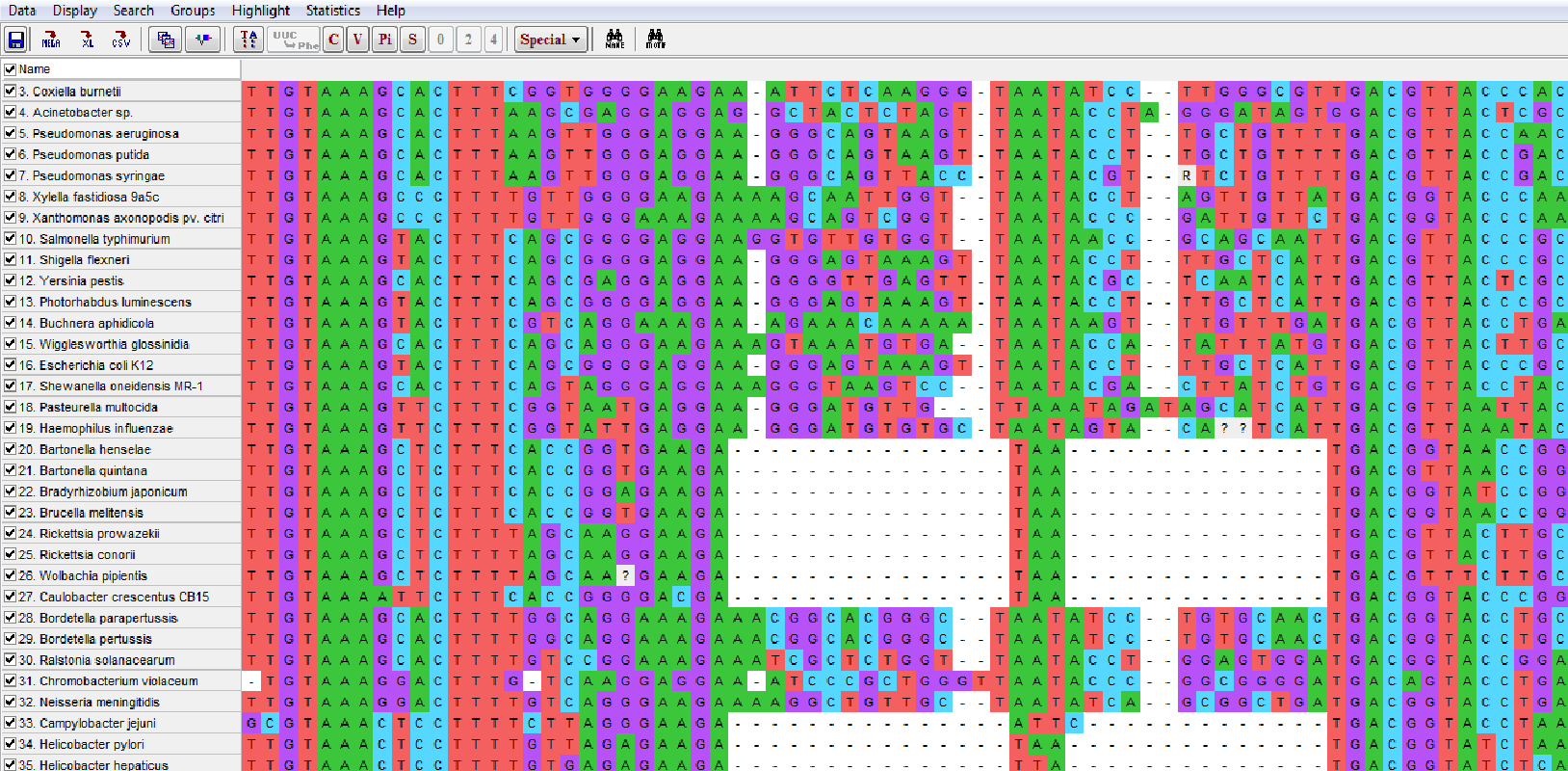
顶部的 C、V、Pi 和 S 按钮允许你根据保守性、变异性、系统发育("简约")信息或单一变化(仅在一条序列中出现)高亮特定位点。简约信息性(Pi)位点在至少两条序列中具有与其他序列不同的核苷酸。这些位点有潜力构建至少最小的分支——两个末端单元。每个类别的计数显示在屏幕左下角。点击 TA 和 C 按钮可以在整个比对中高亮保守位点。
你将看到比对中有一些间隙,只有一条或少数序列存在。此比对不包含外群序列,因此原则上我们只能产生无根树。
问题:
- 比对中的间隙代表什么?
- 此比对中系统发育信息性(Pi)位点的百分比是多少?
- 我们如何尝试为我们比对找到合适的外群序列?
- 鉴于我们的树将是无根的,我们能否仍然推断不同变形菌纲的单系性?
估计序列分歧度:成对距离,30 分钟
现在关闭 Data Explorer,前往 Distance,计算比对中所有序列之间的成对距离比较。选择 Compute Pairwise Distances。首先通过选择 p-difference(即任意两条序列之间差异位点的比例,类似于你在第 2 章中看到的 % 一致性得分)计算原始的、未校正的距离度量。点击 Analysis Preferences 窗口中 Setting 部分(Option 部分旁边)的区域,使用以下设置:
- 方差估计方法 = None。
- 模型/方法 = p 距离。
- 包含的替换 = 转换 + 颠换。
- 位点间速率 = 均匀速率。
- 缺口/缺失数据处理 = 成对删除。
并点击 Ok。
从 Average 菜单中选择 Overall。这些序列之间的总体平均序列分歧度是多少?
现在重复上述步骤,但这次选择 Kimura 2 parameter(K2P)模型。现在的总体平均距离是多少?(始终使用 Caption 菜单选项验证你的设置)。
我们在 Data Explorer 中看到,比对中有高度保守但也有高度变异的位点。假设均匀速率可能不现实。重复上述步骤,这次同时选择 K2P 模型和 Rates among sites = Gamma distributed(G)。现在的总体平均距离是多少?
实际上,我们还希望序列中的碱基组成能够不同于到目前为止假设的 1:1:1:1。因此,保持所有其他设置与你之前的设置相同,现在在 Model/Method 下选择 Tamura-Nei(TN)模型并计算总体平均距离。
最后,你会回忆起比对中有一些大的间隙。因此,重复上一步,但这次将 Gaps/Missing Data Treatment 更改为 Complete deletion。现在的总体平均距离是多少?
问题:
- 为什么这些距离值越来越大(除了最后一步)?
- 你能想到哪些情况下原始 p 差异可以用于测量进化距离?
- Complete deletion 是什么(检查你的 Captions),它与 Pairwise deletion 相比如何?
- 在你看来,对这组序列进行序列分歧度建模的最佳方式是什么?
从序列分歧度到树:距离树,60 分钟
在计算了序列之间的**成对距离**后,你现在将进行**距离树**分析(NJ),将成对序列距离转换为(距离)树。
使用 MEGA 11,在 Phylogeny 菜单下构建两棵邻接树:一棵基于 p 差异并使用 Uniform rates("Rates among Sites")、Complete deletion("Gaps/Missing Data Treatment"),另一棵基于你认为的最佳模型/处理(根据作业 III 问题 4 的回答),包括"Gamma Distributed"速率。你能看到(树)结果有差异吗?
现在重复最佳模型 + Gamma 分析,这次使用 Pairwise deletion(而非 Complete deletion)。应该有一个 Heliobacter/Campylobacter + Desulfo/Geobacter Bdellovibrio 分支。你可以通过 I) 点击其茎部分支,II) 使用 Subtree > Root tree 来用此分支作为根。检查你的树拓扑结构是否发生了变化。
在同一棵树上,在 Tree Explorer 中使用 Options 按钮(窗口顶部,i 按钮左侧)显示树格式化工具,用于让你的两棵树看起来漂亮。(你也可以使用 TaxonNames > ShowTaxonMarkers > Markers)。使用 Tree、Branch 和 Labels 标签并探索各种选项。例如,更改树的外观,按需切换分支长度的显示,并使用 Labels 标签为 α、β、γ、δ 和 ε 变形菌标记各自的颜色和形状(示例见上图中作业 II 的图)。使用 Image > Save as PNG file 保存树的图像。
记住此分析实际上是无根的,因此也要通过从 View > Tree/Branch Style 选择 Radiation 产生无根的可视化。这是完全相同的树结构和分支长度,但它没有被"极化",因此其中分支的存在取决于根的选择。
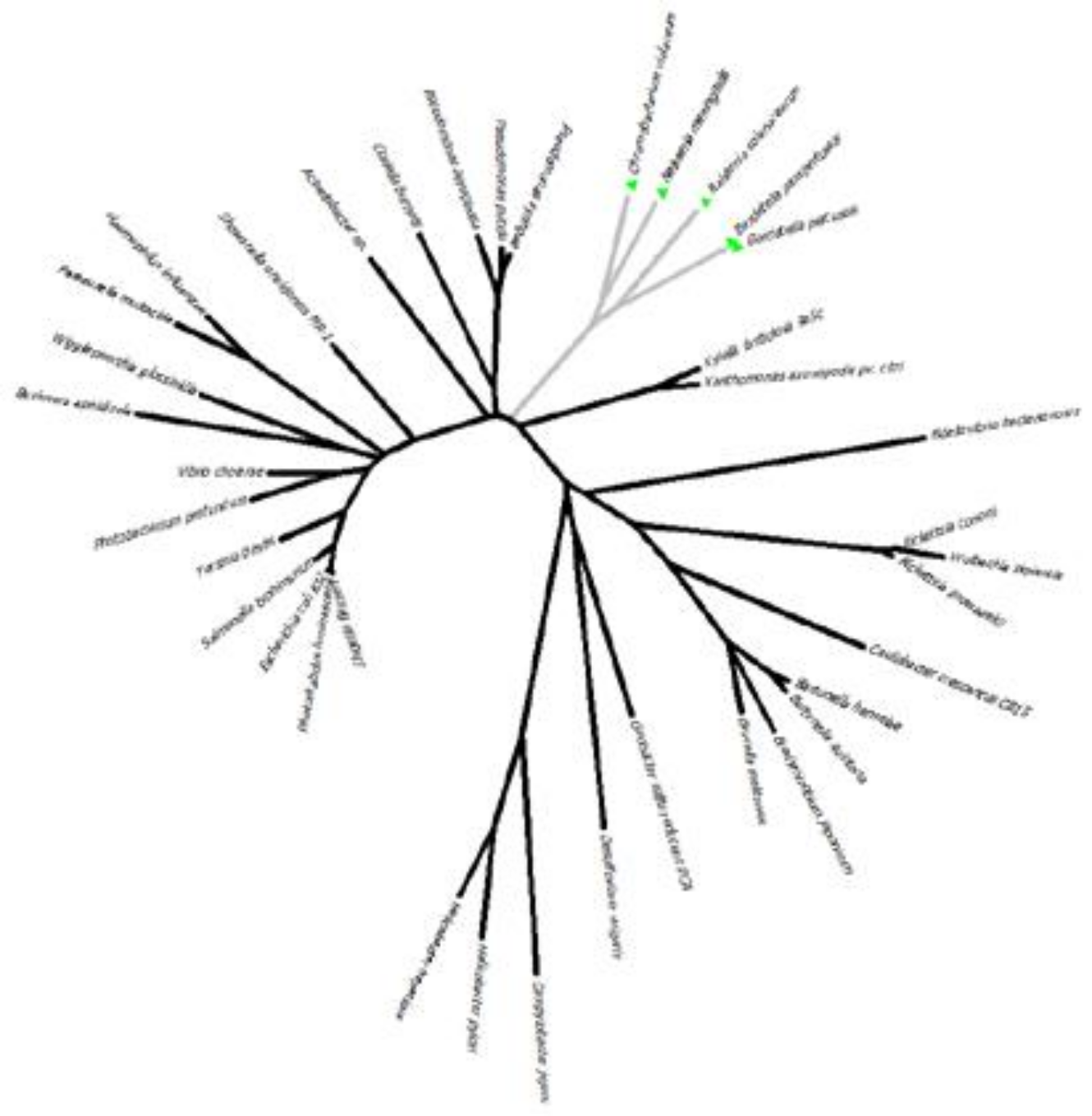
问题: 1. 记住对根选择的依赖性,根据此序列比对,你能对 α-、β-、γ-、δ- 和 ε-变形菌的单系性说什么? 2. 按上述根化(将 Bdellovibrio 和 Heliobacter 作为外群),你的 NJ 树与图中描绘的一致吗?如果不一致,可能是什么原因?
从序列分歧度到树:似然树,60 分钟
使用最大似然(ML)方法,应用显式的核苷酸替换模型以计算"观测数据"的概率。这意味着给定某个树拓扑结构(在许多此类拓扑结构和显式核苷酸替换模型中)计算每个位点按观测方式变化的概率。ML 树上的分支长度是速率,以每个位点的核苷酸替换数表示。
你将首先使用 MEGA 计算 ML 树,然后将其与常用的在线工具 IQ-TREE 进行比较。
在 MEGA 中使用 Phylogeny 按钮选择 Construct/test Maximum Likelihood Tree 并使用以下设置计算树:
- 系统发育检验 = None。
- 模型/方法 = General Time Reversible model。
- 位点间速率 = Gamma Distributed (G)。
- 缺口/缺失数据处理 = Complete deletion。
- ML 启发式方法 = SPR level 5。
- ML 初始树 = NeighborJoining。
- 分支交换过滤器 = None。
ML 启发式方法:在 ML 搜索期间用于从一个树"跳"到另一个树的分支交换器;"Initial tree for ML":此搜索的起始树。
分析完成后(通常几分钟),让你的 ML 树看起来漂亮并产生无根版本(如你之前所做的),并检查 Caption 中获得的 log 似然值。你可能想取消选择"Branch Lengths"。五个变形菌纲在此分析中如何分组?ML 树拓扑结构与你在作业 IV 开始时计算的 NJ 树不同吗?
作为比较,现在前往 IQ-TREE,你将看到三个模块:
"Tree Inference"、"Model Selection" 和 "Analysis Result"。选择"Tree Inference",只需上传你的原始变形菌 SSU 序列比对 FASTA 文件,目前只使用默认选项,即不要更改任何内容。
IQ-TREE 很可能是目前计算 ML 系统发育树及其关联 Bootstrap 分析最好、最强大的工具。如有延迟或分析时间过长,你也可以使用 IQ-TREE webserver 并选择 Vienna 和/或 Los Alamos(页面顶部列出的 3 个 Web 服务器之一,CIPRES 是付费站点)。
取决于服务器负载(见左上角),在令人惊讶的短时间(几分钟?)后,在 Analysis Results > Run Log 中检查你的状态,持续刷新屏幕。当分析完成后,立即前往左下角的 DOWNLOAD SELECTED JOBS 按钮,将结果 .zip 解压到你自己的空间。在 Summary 标签中你已经可以看到你的树结果,节点上标有实际的 Bootstrap 支持值。在 Analysis Result 标签中你可以看到类似以下内容:
ModelFinder
-----------
Best-fit model according to BIC: TN+I+G4
List of models sorted by BIC scores:
Model LogL AIC w-AIC AICc w-AICc BIC w-BIC
TN+I+G4 -19303.1910 38766.3819 + 0.0989 38771.8665 + 0.1070 39230.4932 + 0.6952
TIM3+I+G4 -19300.2294 38762.4589 + 0.7035 38768.0829 + 0.7096 39232.3716 + 0.2718
TIM2+I+G4 -19303.0277 38768.0554 + 0.0429 38773.6795 + 0.0432 39237.9681 - 0.0166
TIM+I+G4 -19303.2163 38768.4327 - 0.0355 38774.0567 - 0.0358 39238.3454 - 0.0137
HKY+I+G4 -19313.5323 38785.0647 - 0.0000 38790.4115 - 0.0000 39243.3746 - 0.0011
TPM3+I+G4 -19310.1240 38780.2479 - 0.0001 38785.7325 - 0.0001 39244.3592 - 0.0007
TPM3u+I+G4 -19310.1375 38780.2751 - 0.0001 38785.7596 - 0.0001 39244.3864 - 0.0007
GTR+I+G4 -19300.0064 38766.0128 + 0.1190 38771.9213 + 0.1041 39247.5283 - 0.0001
TPM2u+I+G4 -19313.1016 38786.2031 - 0.0000 38791.6877 - 0.0000 39250.3144 - 0.0000
TPM2+I+G4 -19313.1024 38786.2047 - 0.0000 38791.6893 - 0.0000 39250.3160 - 0.0000
K3Pu+I+G4 -19326.9125 40011.8250 - 0.0000 40017.1719 - 0.0000 40470.1349 - 0.0000
TVMe+I+G4 -19336.2200 40028.4401 - 0.0000 40033.6511 - 0.0000 40480.9486 - 0.0000
SYM+I+G4 -19332.9153 40023.8306 - 0.0000 40029.1775 - 0.0000 40482.1405 - 0.0000
K2P+I+G4 -19348.9891 40047.9782 - 0.0000 40052.7924 - 0.0000 40483.0826 - 0.0000
TNe+I+G4 -19345.7611 40043.5223 - 0.0000 40048.4669 - 0.0000 40484.4280 - 0.0000
K3P+I+G4 -19348.6125 40049.2250 - 0.0000 40054.1697 - 0.0000 40490.1308 - 0.0000
TIME+I+G4 -19345.4002 40044.8003 - 0.0000 40049.8772 - 0.0000 40491.5074 - 0.0000
TIM2e+I+G4 -19345.7324 40045.4647 - 0.0000 40050.5417 - 0.0000 40492.1719 - 0.0000
F81+I+G4 -20192.9782 40539.9564 - 0.0000 40545.0334 - 0.0000 40986.6636 - 0.0000
JC+I+G4 -19637.5080 39425.0160 - 0.0000 39429.8302 - 0.0000 39860.1204 - 0.0000
AIC, w-AIC : Akaike information criterion scores and weights.
AICc, w-AICc : Corrected AIC scores and weights.
BIC, w-BIC : Bayesian information criterion scores and weights.
这些是此数据集与第一列中指示的每个核苷酸替换模型组合的 LogL 值,即"log 似然值"。
LogL 值都是负数,因为 log 函数在 0 和 1 之间是负的,而似然值(通常是概率的乘积)也是。对于这些值的计数:越少负数越好。因此,最佳拟合模型标在最上面,LogL = -19303.1910,而最差拟合模型(1 参数 Jukes Cantor 模型)的 LogL = -21977.4853。
LogL 的差异是否"显著"由其他列中的统计量衡量:"Akaike Information Content"(AIC)和"Bayesian Information Content"(BIC)。使用 BIC 作为准则,IQ-TREE 决定 TN+I+G4 模型最适合数据。参见 IQ-TREE 文档了解模型解释。
到目前为止这是模型选择,接下来是实际的树搜索和数据的最大似然估计。打开你从 IQ-TREE 下载的 .zip 包中的 .log 文件,在完成树搜索后找到"Best score found"。它如何与模型测试的最佳 Ln 得分比较?你能解释这个差异吗?
现在将你的 IQ-TREE 分析的最佳 LogL 得分与之前在 MEGA 11 中找到的最佳 log 似然值进行比较。它们相同吗?尝试在下方标记为'o'的单元格中找到 LnL 值。
| 模型 | MEGA 成对删除 | MEGA 完全删除 | MEGA 使用所有位点 | IQ-TREE |
|---|---|---|---|---|
| TN+I+G | o | -11895.49 | o | o |
| GTR+G | -13847.07 | o | -19330.23 | o |
问题:
- 为什么 LnL 值在不同特征集(删除处理)之间差异如此之大,但在不同模型之间差异不大?
- 为什么我们不直接使用最复杂的模型,它肯定最适合任何数据集?
为了在 iTOL 中可视化你的带 Bootstrap 值的 IQ-TREE ML 树,使用作业 I 中解释的方法。前往 iTOL,打开并上传你从 IQ-TREE Web 服务器下载的 .zip 文件中的 .contree 文件。扩展名"contree"指的是你正在处理的是 Bootstrap 一致树,即 100 棵 Bootstrap 树的 50% 多数规则汇总。
制作一张漂亮的树图,确保方向和(无)根化一致,然后你可以通过以 .svg 图形格式导出来保存。将此与上图中作业 II 的图进行比较,讨论你可能看到的任何差异,包括拓扑结构和节点支持度。
可选:树与 MSA 一样好:TDP DNA 序列的系统发育估计,60 分钟
此作业的目的是学习如何使用 Mesquite 软件包在系统发育分析之前处理和操作 DNA 序列比对。Mesquite(实际上)不建树,但允许你优化比对、编辑特征矩阵、在系统发育树上追踪特征,并且非常有用地,在令人印象深刻的文件格式列表之间转换。Mesquite 是免费软件,可以从这里获取,附带工具的有用描述。
TDP 依赖的酶超家族
为酶家族构建基因树有助于识别酶功能如何进化。思路是生成一棵基因树,包含来自广泛分类单元的功能和结构相关序列,然后 I) 尝试预测基因家族中尚未被实验表征的成员的功能,II) 推断酶功能本身在整个超家族中的进化,包括重建祖先状态。显然,这些问题只能从系统发育角度解决。
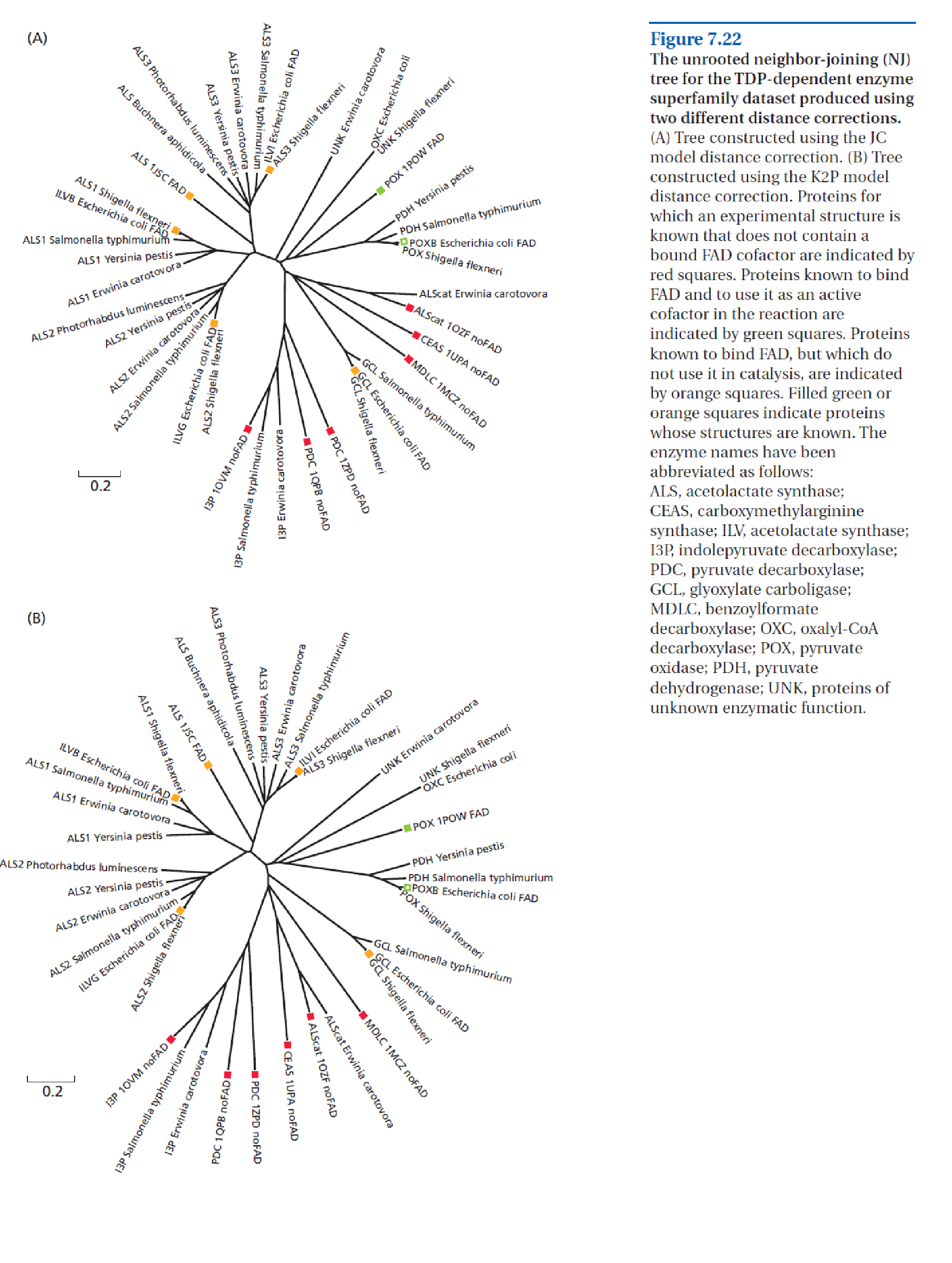
我们今天用来展示此方法的序列比对包含 38 个 γ-变形菌序列(源自 [@bioinformatics_2007]),来自大肠杆菌、沙门氏菌及相关物种等知名肠道菌。这些序列代表了一系列酶功能,描述于下图中。
现在的主要问题是如何获得准确的多重序列比对,这确实是本实践的重点。虽然没有明确说明,Zv&B 给出的比对可能是使用 CLUSTAL 构建的,这是一种基于引导树的渐进式比对方法。虽然这种方法快速且强大,但在比对区域中序列保守水平不同的情况下存在已知问题。
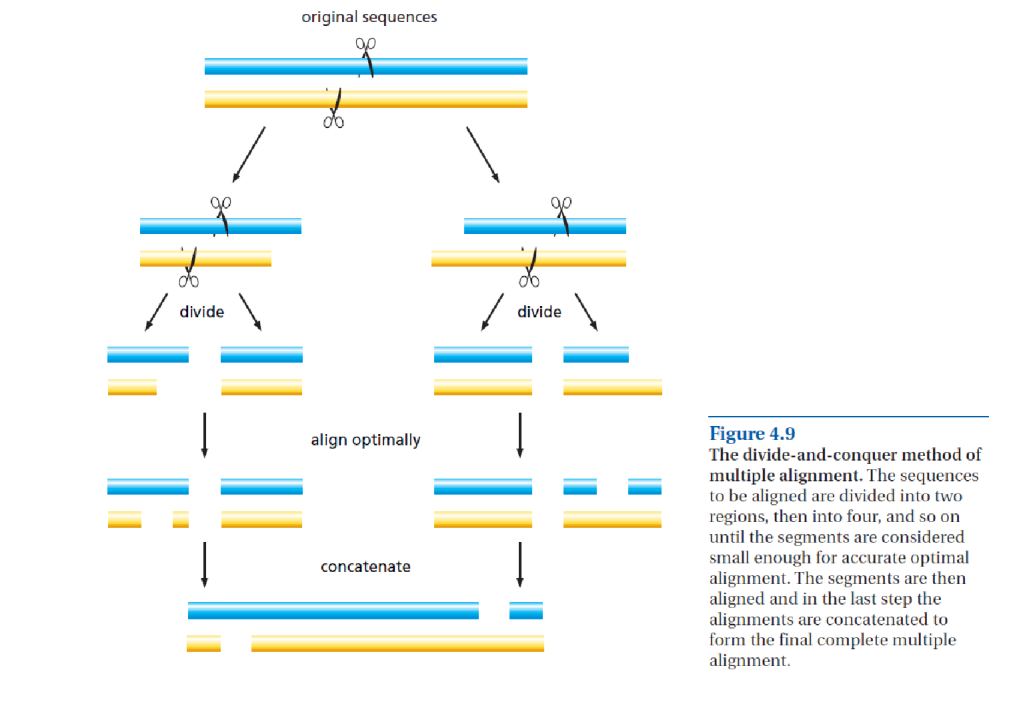
因此,在本作业中,除了重现下图中描述的模式外,你将使用 MAFFT 重新比对序列矩阵,随后进行系统发育分析和解读。MAFFT 采用基于 k-mer 的方法,允许在比对前将序列"拆分"为高保守和变异部分,然后在比对后将"已比对的部分粘合在一起",即分治法(divide-and-conquer approach)。
任务: 比对与 MAFFT 重新比对
- 分析 TDP 依赖的酶超家族序列矩阵(名为
TDP_dependent_enzyme_DNA.fas,可从 Brightspace 获取),方法类似于你在上一个实践中分析变形菌 16S rRNA 序列比对的方式。基本上,使用TDP_dependent_enzyme_DNA.fas执行简约分析并使用 IQ-TREE 生成最大似然树。 - 要生成 TDP 序列矩阵的替代版本,前往 MAFFT 并在那里上传你的 TDP .fas 文件。
- 查看所有可用选项但使用默认设置,即不要更改任何设置。点击
Submit并等待,大约一分钟。 - 你将看到一些分析进度信息以及表示所有序列共线性的点图。根据服务器负载,点击
Check now链接将返回你的重新比对序列。 - 点击
Reformat,选择 PAUP|NEXUS 作为输出序列格式,然后选择Download to file并点击Submit。 - 现在启动你刚刚在笔记本电脑上安装的 Mesquite。准备好后,打开你刚从 MAFFT 服务器下载的文件。点击
Display > Widths > Thin Rows并选择Display > Bird's Eye View以获得更好的比对视图。 - 你现在需要在这些序列中推断阅读框,即第 1、第 2 和第 3 密码子位置是什么。我们这样做:
- 点击
Characters > List of characters。 - 按 Ctrl-A 选择所有位点。
- 选择列标题
Codon position,然后Set codon position > Minimise stop codons。 - 返回 Matrix 标签。
- 序列比对现在按密码子结构化;为了更清楚地可视化,选择
Display > Color Matrix Cells > Color nucleotide by amino acid。 - 点击
Display > Show Color Legend以解读氨基酸级别的模式。理想情况下,不应有或只有少量终止密码子(黑色)出现。 - 以合理的名称保存此文件,并给它
.nex扩展名(nexus 文件格式)。同时选择File > Save Matrix as PDF并保存到你自己的空间。 - 选择
File > Export > Simplified NEXUS以便此 MAFFT 比对的矩阵可以导入 IQ-TREE。
任务: 比较原始与 MAFFT IQ-TREE 和分支支持度
现在你有两个版本的 TDP 序列矩阵,可以开始比较树拓扑结果。因此,对 MAFFT 重新比对的矩阵(Simplified NEXUS 格式)执行如上所述的简约分析和 IQ-TREE ML 分析。
- 通过对原始 TDP 序列矩阵再次执行任务 6-9(上一页)来比较实际 MSA。如对 MAFFT 重新比对矩阵所做的那样保存矩阵的 .pdf。通过"目视检查"比较两个 .pdf,讨论你认为哪一个"更好"。
- 确保比较原始 IQ-TREE 与 MAFFT 重新比对分析的 IQ-TREE,并讨论差异。
- 你会注意到上图中没有给出 Bootstrap 支持值,但现在你在 IQ-TREE 中有了。解读 IQ-TREE 中的 Bootstrap 分支支持度,并讨论 Zvelebil & Baum 关于功能进化的说法是否得到支持,以及我们在多大程度上可以对此有信心。
原始: 
MAFFT: 
原始,IQ-TREE: 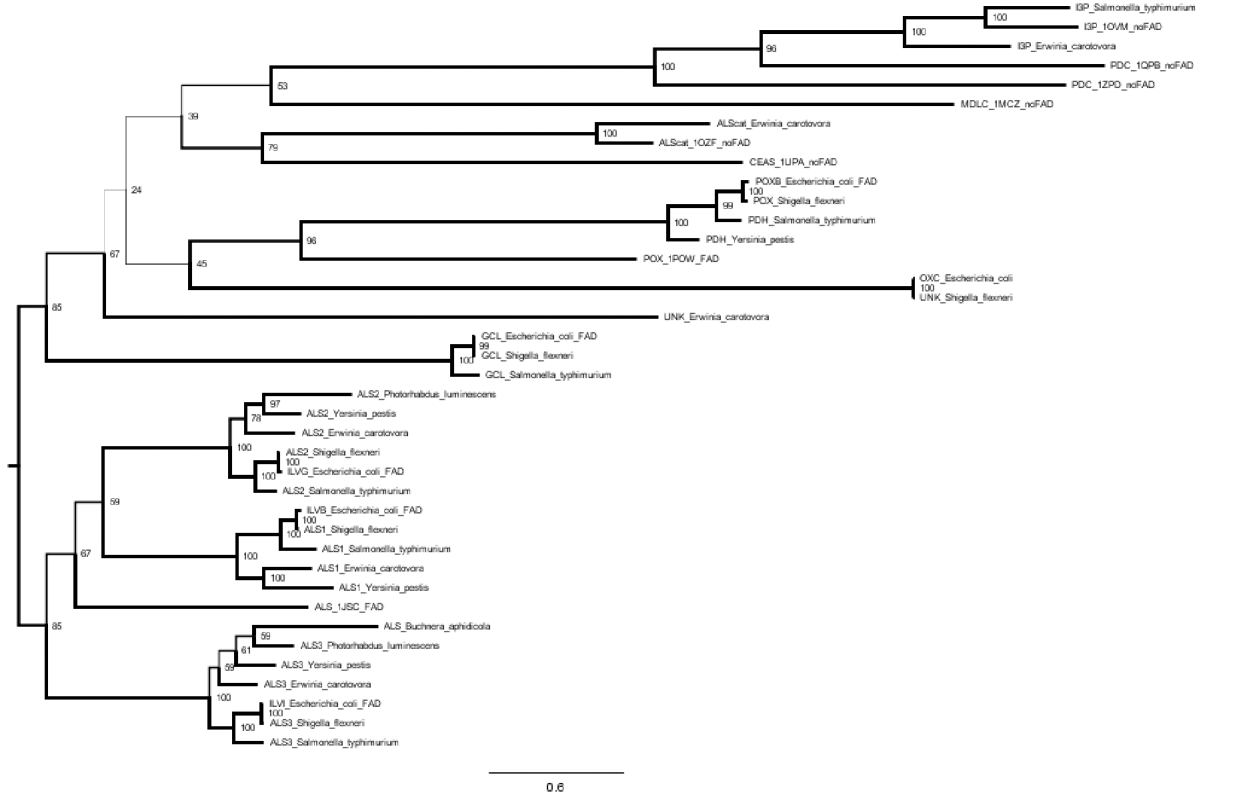
MAFFT 比对,IQ-TREE: 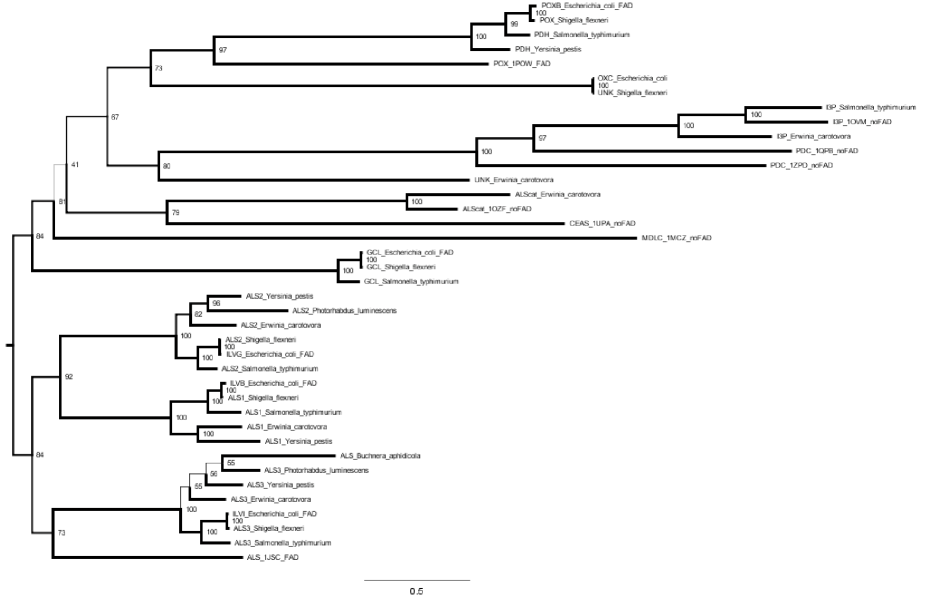
项目准备练习
使用上一章的 ARF 多重序列比对(氨基酸序列),在 MAFFT 中对齐,现在使用 MEGA 11 构建 NJ 树,分别使用 Poisson 和 Dayhoff 氨基酸替换模型。
使用 FigTree 制作合适的树图;然后在 IQ-TREE 中生成最大似然树(带 Bootstrap),并在 FigTree 中制作其漂亮图片。
树是否正确根化?如果尚未包含外群,可以添加什么外群?
用几个要点描述以下各项。你可以包含最多两个图或表。
- 材料与方法 你做了什么?你使用了哪些数据、数据库和工具,为什么选择它们?你选择了哪些重要设置?
- 结果 你发现了什么,主要结果是什么?报告相关数据、数字、表/图,并清楚描述你的观察。
- 讨论与结论 结果合理吗?是否符合你的预期,还是你看到了令人惊讶的东西?结果意味着什么,你如何解读它们?不同工具是否一致?你能得出什么结论?确保描述你解读背后的预期和假设。
参考文献¶
{{ bibliography }}
术语表¶
- 加性树(Additive tree)
- 分支长度与数据变化量成正比的系统发育树。
- 自衍征(Autapomorphy)
- 唯一衍生的特征状态。
- 贝叶斯推断(Bayesian inference)
- 一种基于特征的系统发育方法,在给定模型和数据下估计树的后验概率。
- 二叉的(Bifurcating)
- 一种树,所有节点都通过三个分支连接。
- Bootstrap
- 一种衡量系统发育树中节点支持度的方法。
- Bootstrap 值(Bootstrap value)
- 分支(clade)在 Bootstrap 重复树中出现的群频率(百分比)。
- 分支长度(Branch length)
- 加性系统发育树中分支的长度,以步(简约)、距离(聚类)或每位点替换数(最大似然)表示。
- 特征(Characters)
- 生物体的可观察特征或性状。在分子系统发育中,序列比对(MSA)中的每个位置被视为一个特征。
- 基于特征的方法(Character-based methods)
- 直接评估位点模式的树重建方法(如简约法、ML、贝叶斯)。
- 分支(Clade)
- 单系群,MRCA 及其所有后代。
- 分支图(Cladogram)
- 仅显示拓扑结构的树图;分支长度不与变化成正比。
- 一致树(Consensus tree)
- 一组同等好的树的汇总树(如严格一致树、50% 多数规则一致树)。
- 基于距离的方法(Distance-based methods)
- 基于成对序列距离的树重建(如邻接法)。
- 基因复制(Gene duplication)
- 在谱系中产生一个基因额外拷贝的事件。
- 基因丢失(Gene loss)
- 基因拷贝从谱系中消失。
- 基因树(Gene tree)
- 从基因(或蛋白质)序列重建的系统发育树,可能包含旁系同源物。
- GTR
- 广时间可逆核苷酸替换模型。
- HTU
- 假想分类单元;代表推断祖先的内部节点。
- JC
- Jukes-Cantor 核苷酸替换模型。
- K2P
- Kimura 2 参数核苷酸替换模型。
- LBA
- 长枝吸引(Long-branch attraction)。
- 最大似然(Maximum likelihood, ML)
- 一种基于特征的方法,选择使观测数据概率最大化的树和模型参数。
- MLE
- 最大似然估计(Maximum likelihood estimate)。
- 单系性(Monophyly)
- 一个群体的所有成员都源自一个 MRCA。
- MRCA
- 最近共同祖先(Most recent common ancestor)。
- 邻接法(Neighbor Joining, NJ)
- 一种基于距离的建树算法,通过迭代合并邻居来最小化总树长。
- Newick
- 一种用于编码树拓扑(可选分支长度)的括号文本格式。
- 节点支持度(Nodal support)
- 数据对系统发育树中节点的支持(通常用 Bootstrap 值衡量)。
- OTU
- 操作分类单元(Operational taxonomic unit)——用于分类密切相关的个体群。
- 外群(Outgroup)
- 感兴趣群体之外的参考分类单元,用于根化树。
- 并系性(Paraphyly)
- 一个群体的并非所有成员都源自一个 MRCA。
- p 差异(p-difference)
- 比例差异,未校正。
- 系统发育学(Phylogenetics)
- 研究生物或基因之间的进化历史和关系。
- 多分支(Polytomies)
- 系统发育树中塌缩的部分;节点通过超过三个分支连接。
- 协调树(Reconciled tree)
- 通过添加复制和丢失事件来解释不一致性,将基因树与物种树一起解读。
- 根(Root)
- 用于极化树的参考分类单元或节点。
- 根化(Rooting)
- 为树指定根以推断进化方向的行为。
- 姐妹群(Sister group)
- 两个彼此共享独占 MRCA 的分支。
- 物种树(Species tree)
- 概括物种关系的系统发育树,可能由多个基因树信息支持。
- 状态(States)
- 特征在给定个体或分类单元中的特定条件或表现形式。在分子系统发育中,核苷酸 MSA 中每个位置(特征)的状态可以是 A、T、C 或 G。
- 共同衍征(Synapomorphy)
- 共享衍生的特征状态。
- 性状(Trait)
- 生物体的独特表型特征,受遗传和环境因素影响。
- 转换(Transition)
- 嘧啶之间(C↔T)或嘌呤之间(A↔G)的替换。
- 颠换(Transversion)
- 嘧啶 ↔ 嘌呤的替换。
- 树拓扑结构(Tree topology)
- 系统发育树的分支结构,与分支长度无关。
- 超度量不等式(Ultrametric inequality)
- 在任意三元组中,两个最大成对距离相等。
- 超度量树(Ultrametric tree)
- 一种有根树,从根到末端的所有路径长度相等(可用时间单位解释)。
- 三角不等式(Triangle inequality)
- 对于任意三角形,边长为 \(a\)、\(b\) 和 \(c\),任意两边之和必须大于或等于第三边: \(a \le b + c; b \le a + c; c \le a + b\)
本站总访问量 次