第二章
课程来源与翻译声明
- 原著版权:本教程翻译自瓦赫宁根大学(Wageningen University & Research)的经典开源课程 Introduction to Bioinformatics。
- 翻译说明:本版本主要采用人工智能(机器翻译)进行全文本地化,并由团队进行了初步校对。部分专业术语可能存在翻译不够地道或准确的地方。
- 勘误反馈:若你在学习过程中遇到语病、错别字、代码失效或概念歧义,欢迎随时点击页面右上角的 :material-edit: 编辑此页(或联系课题组/在 GitHub Issue 中)提交反馈,帮助我们持续完善本教程。
在本章中,你将学习序列比对的相关知识。 在实践环节中,你将学习如何进行双序列比对和多序列比对、执行序列搜索和模体分析、设计引物,并讨论实验结果。
学习目标
学习本章后,你应该能够: - 描述 DNA 和蛋白质比对的用途 - 解释局部比对与全局比对的区别 - 描述与比对和序列搜索相关的概念,如点阵图 (dotplot)、比对得分、E 值和替换矩阵 - 解释多序列比对的用途以及不同多序列比对解决方案之间的差异 - 描述模体是什么以及概隐马尔可夫模型的基础知识 - 设计 PCR 引物并描述必要的考虑因素
引言¶
序列比较是应用生物信息学领域的关键工具。 通过分析 DNA 和蛋白质序列,研究人员可以对基因组中新基因进行功能注释、构建蛋白质结构模型,并研究基因表达。 值得注意的是,自然界倾向于保留有效的机制,而不是为每个物种重新发明轮子。 生物体从祖先进化而来并不断积累突变。 这里我们讨论的是影响少量碱基的**小规模**突变:替换(另见第一章)以及小的插入和缺失。
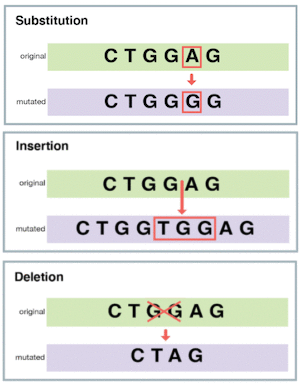
不同类型的小规模突变。来源:https://evolution.berkeley.edu/,CC BY-NC-SA 4.0。
通过突变,生物体可以随着时间的推移逐渐发展出新的性状。 这些进化关系也意味着可以在不同生物体中找到相似的基因,如果两个蛋白质具有一定的相似性,功能注释可以从一个蛋白质转移到另一个。 然而,即使两个蛋白质看起来相似,它们也可能具有不同的功能。 一般来说,相似性源于共同的祖先(趋异进化),但相似性也可能独立出现(趋同进化)。 在深入分析序列是否相关之前,理解一些关键术语非常重要。
同源性与相似性
同源性**意味着序列具有共同的进化历史,因此拥有共同的祖先。 同源性是不可量化的。 如果两个序列拥有共同的祖先,它们就是**同源的。 因此,两个序列要么同源,要么不同源。
**序列一致性**和**序列相似性**通常被用来推断两个序列是否同源。 我们可以测量序列之间的一致性或相似性,我们将在本章后面看到如何进行这些操作。
相比之下,我们无法测量同源性,只能推断它。
延伸阅读
这是一篇经典的论文,介绍了同源蛋白质家族:Tatusov et al., 1997。
本章涵盖序列比较的基础知识。我们将描述如何使用点阵图或双序列比对来比较两条序列。 然后,介绍在数据库中搜索相似序列的方法。 涵盖比较多条序列的不同方法:用于对齐的多序列比对、用于发现共有序列的模体以及用于表示多条序列的概隐马尔可夫模型。 本章最后以 PCR 引物设计为示例,介绍序列比对算法在实际中的应用。
点阵图 (Dot plots)¶
点阵图是一种可视化两条序列之间相似区域的简单方法。 它以矩阵形式表示,其中一条序列纵向书写,另一条横向书写。 在残基相同的单元格中放置一个点。 在生成的图中,相似区域显示为对角线段,而插入和缺失显示为对角线上的不连续处。 序列也可以与自身进行比较,此时主对角线将充满点,额外的重复序列出现在偏离主对角线的位置。

一个小型的点阵图示例。来源:CC BY-NC 4.0 [@own_2_2024]。

一个用于将序列与自身比较的点阵图示例。来源:CC BY-NC 4.0 [@own_2_2024]。
这种标记相同残基的简单方式会带来大量的背景噪音。 为了检测有意义的模式,通常会应用过滤器。 例如,在一定的窗口大小(即连续考虑的残基数)内需要满足最小一致性。 这一功能已在一个用于可视化点阵图的网络服务器 dotlet [@dotlet_2000] 中实现。
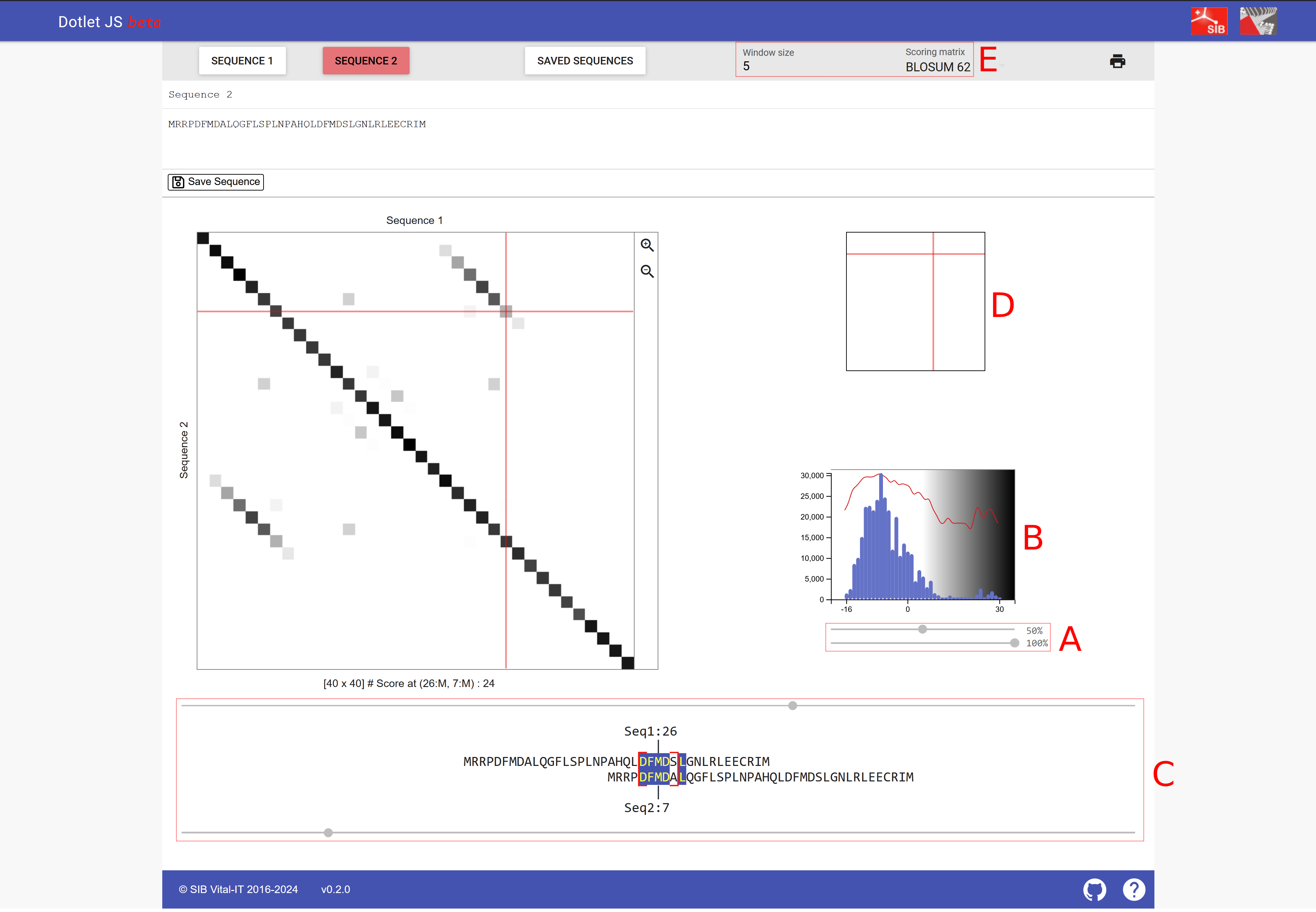
dotlet 的截图,以下蛋白质序列被提交为序列 1 和序列 2:MRRPDFMDALQGFLSPLNPAHQLDFMDSLGNLRLEECRIM。
- (A) 两个滑块用于调整图表的外观:顶部滑块可以调节灵敏度,向右移动则显示的相似区域更少;向左移动则也会显示相似性较低的区域。 底部滑块调整颜色方案,与顶部滑块相比重要性较低。
- (B) 直方图指示显示了特定相似性的命中数量;因此滑块可以调整到直方图的右尾部分。
- (C) 两个滑块可以调整两条序列相互之间的位置。
- (D) 功能类似于 C 的两个滑块,但允许使用方向键导航点阵图。
- (E) 在这里可以选择序列比较的窗口大小和评分矩阵(窗口大小在下文解释,替换矩阵也在下文解释)。
来源:[@dotlet_2000]。
双序列比对 (Pairwise alignment)¶
点阵图提供了一种可视化比较两条序列的方式,但不能提供两条序列之间的相似度。 要计算序列相似性或序列一致性,我们需要执行**双序列比对 (pairwise sequence alignment)。 在比对中,两条序列上下排列,可以引入空位 (gap) 来表示残基的插入或缺失。 我们也说两条序列将被**比对。 比对结果包含匹配、错配和空位。
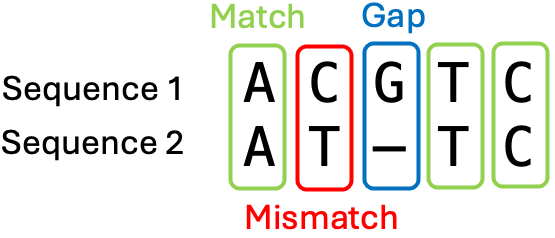
两条比对序列的小型示例。来源:CC BY-NC 4.0 [@own_2_2024]。
DNA 序列的比对¶
序列中的每个位置都可能有插入或缺失,因此空位有许多可能的位置和组合,因此存在许多潜在的比对方案。 最终的比对将是具有最大比对总得分的那一个。 该得分由所谓的_评分参数_决定,这些参数在比对计算之前选定。 DNA 序列评分参数的一个例子是:匹配得分为 1,错配得分为 -1,空位罚分为 -1。 比对的**总得分**是通过对所有列的得分求和来计算的。 然后我们可以比较不同比对的得分,得分较高的比对表示序列之间_更好的_比对结果。 然而,评分参数的选择会影响哪个比对将获得最大得分。 要理解参数对最终比对的影响,请填写以下表格。
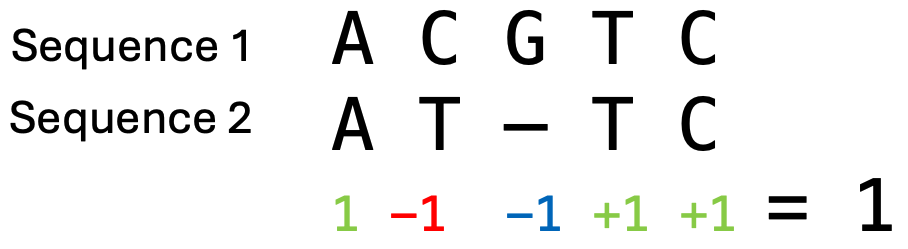
比对得分的计算示例,其中匹配得分为 1,错配得分为 -1,空位罚分为 -1。该比对的总得分为 1。来源:CC BY-NC 4.0 [@own_2_2024]。
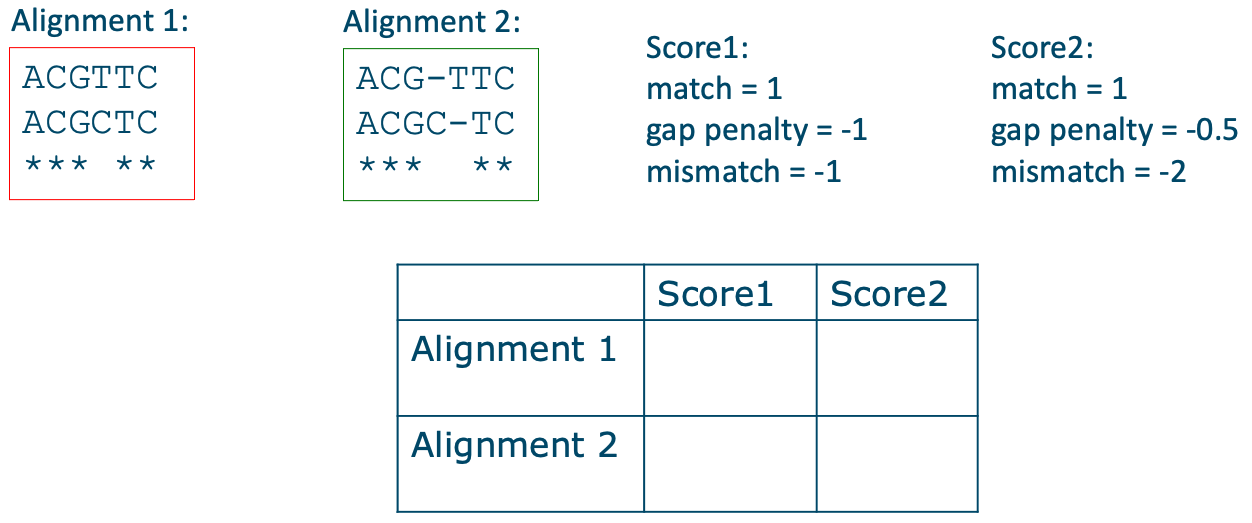
练习: 为两个比对和两组评分参数填写表格。来源:CC BY-NC 4.0 [@own_2_2024]。
注意 2.1:仿射空位代价
我们要比对的 DNA 和蛋白质序列通常具有不同的长度,这是进化过程中插入和缺失的结果。 插入和缺失事件可能影响一个或多个残基,其中长度为 2 的一次事件比两次独立的长度为 1 的事件更有可能发生。 为了在评分方案中考虑这一点,比对程序使用**仿射空位代价 (affine gap costs)**,区分开启空位和延伸空位的代价。 例如,双序列比对程序 needle [@EMBL_tools_2022] 的默认参数为:
空位开启 (gap open,空位中第一个残基的得分):-10
空位延伸 (gap extend,空位中每个额外残基的得分):-0.5
注意 2.2:寻找最佳比对
两条序列可能有大量的比对方案,因为空位可以以许多不同的方式放置。 然而,要找到**最优比对**(即得分最高的比对),并不需要穷举所有可能性。 存在高效的算法可以保证找到最优比对。 Needleman-Wunsch 算法是第一个此类算法,可以在与输入序列长度的平方成正比的时间内解决这个任务。
蛋白质序列的比对¶
替换矩阵¶
在第一章中,我们学习了不同氨基酸具有不同的化学性质。 当蛋白质结构和功能保守时,一个氨基酸更有可能被化学性质相似的氨基酸替换,而不是性质差异很大的氨基酸。 因此,在比对蛋白质序列时,我们希望惩罚化学性质差异较大的氨基酸替换,奖励化学性质相似的替换。 为此,匹配和错配的得分通常由**替换矩阵 (substitution matrix)** 决定,例如 BLOSUM62 —— BLOSUM (BLOck SUbstitution Matrix)。 替换矩阵和空位参数共同决定比对得分。
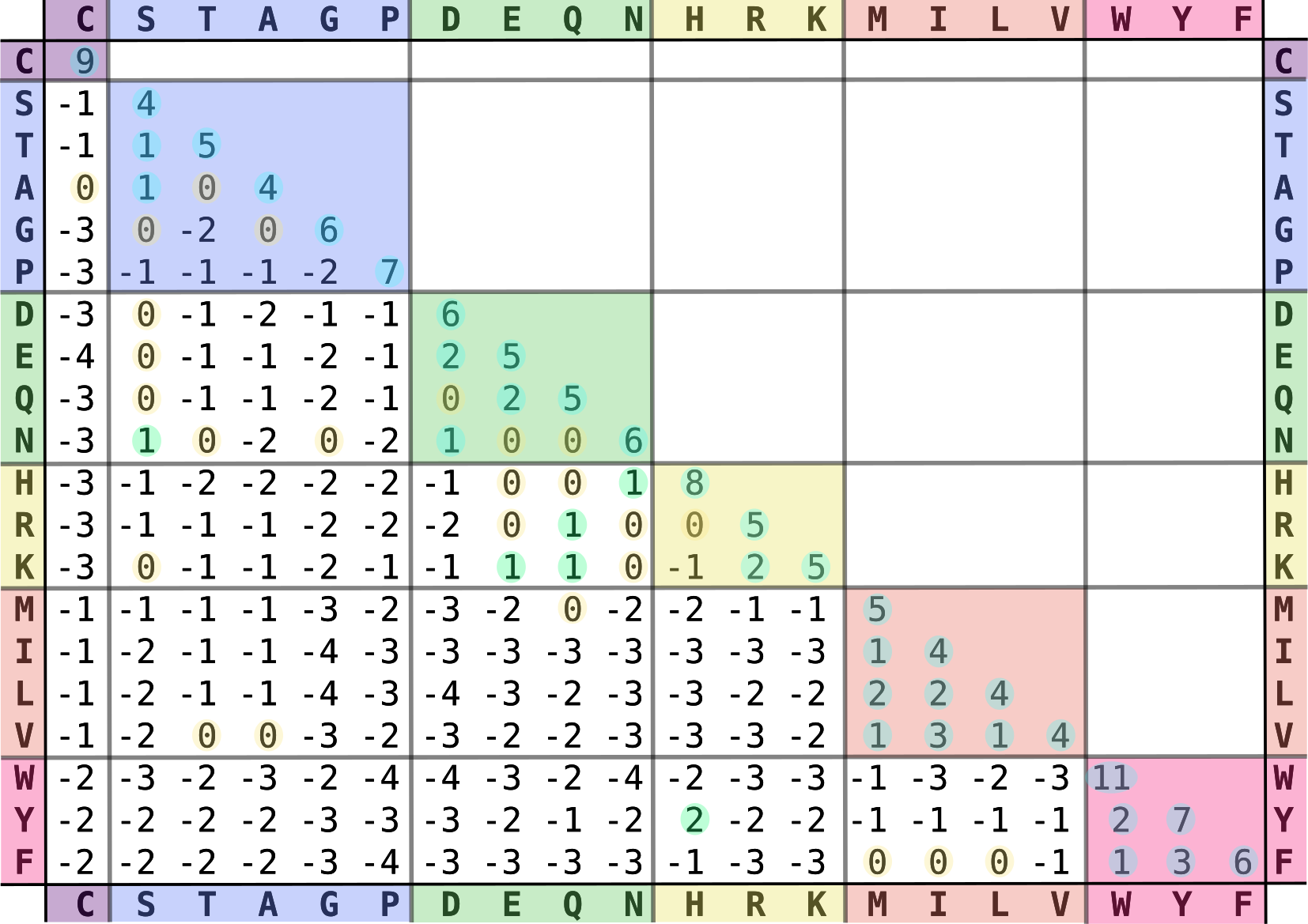
BLOSUM62 氨基酸替换矩阵。矩阵按顺序排列,正值和零值被高亮显示。来源:CC BY-SA 4.0 [@blosum62_2022]。
框 2.1:练习
查看第一章表格中的氨基酸性质,选择一些具有相同性质和不同性质的氨基酸。 然后在 BLOSUM62 矩阵中查找这些氨基酸对。 你观察到了什么?
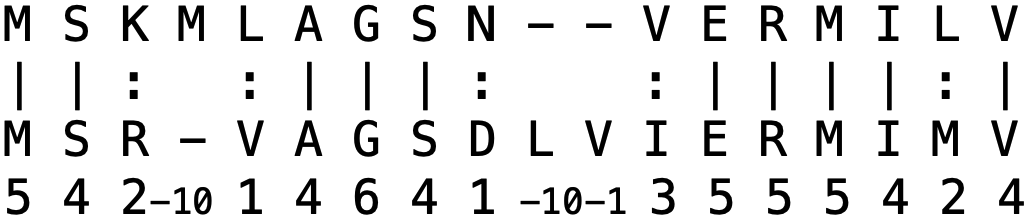
双序列蛋白质比对的示例。使用 BLOSUM62 评分矩阵、空位开启得分 -10 和空位延伸得分 -1,得到的比对得分为 34。来源:CC BY-NC 4.0 [@own_2_2024]。
需要注意的是,我们以氨基酸的化学性质来激励氨基酸替换矩阵的使用;然而,这些性质在确定矩阵时并没有被直接使用。 相反,BLOSUM 矩阵是通过对 Swiss-Prot(第一章)中保守区域进行比对并按一致性进行聚类来确定的。 然后,统计聚类内不同氨基酸对之间的替换情况,用于计算 BLOSUM 得分。 因此,这些得分直接反映了在进化过程中哪些氨基酸更频繁地相互替换,我们可以观察到这种频率与它们的化学性质密切相关。 存在不同版本的 BLOSUM 矩阵;例如,BLOSUM62 是通过将一致性为 62% 的序列进行聚类得到的,适合比较一致性约为 62% 的蛋白质序列。 其他可用矩阵包括 BLOSUM45(用于更趋异的序列)和 BLOSUM80(用于更相似的序列)。
另一类矩阵是 PAM (Point Accepted Mutation),其开发早于 BLOSUM。 PAM 矩阵中的条目表示在特定进化变化单位下氨基酸的替换概率。 例如,PAM1 表示每 100 个氨基酸残基发生一次替换,因此适合非常密切相关的序列。 常用的矩阵是 PAM250,意味着 100 个残基上发生了 250 次突变;也就是说,许多残基受到不止一次突变的影响。

不同可用替换矩阵的概览。来源:CC BY-NC 4.0 [@own_2_2024]。
延伸阅读
PAM 和 BLOSUM 替换矩阵的介绍。
蛋白质一致性与相似性¶
对于两条蛋白质序列,我们可以区分两种不同的相似度度量:一致性和相似性,它们的定义略有不同。 **蛋白质一致性**是相同氨基酸的数量除以比对长度。 **蛋白质相似性**是相似氨基酸数量_和_相同氨基酸数量之和除以比对长度。 在双序列比对程序 needle 中,**相同氨基酸**用竖线 (|) 标记,**相似氨基酸**用冒号 (:) 标记,定义为在所选替换矩阵中具有正得分(即 > 0)的氨基酸对。

蛋白质比对示例。一致性百分比为 10 / 18 = 55.6%,相似性百分比为 14 / 18 = 77.8%。各条序列的长度显示在右侧。来源:CC BY-NC 4.0 [@own_2_2024]。
请注意,双序列比对方法并不直接尝试最大化相似性或一致性,而是通过选定参数产生的最佳比对来计算这些值。 特别是对于进化关系较远的序列,参数对比对结果(从而对估计的一致性和相似性)有很大影响。 以下展示了两个比对,其中来自水稻的同一对蛋白激酶使用了不同参数进行比对。 根据参数的不同,一致性在 17.6% 到 26.2% 之间变化。

使用不同参数对相同两条序列 (LERK1_ORYSI 和 XA21_ORYSI) 的比对: A) BLOSUM62 矩阵,空位开启 -10,空位延伸 -0.5。一致性 = 210/1191 (17.6%),相似性 = 345/1191 (29.0%)。 B) BLOSUM62 矩阵,空位开启 -5,空位延伸 -0.5。一致性 = 305/1166 (26.2%),相似性 = 428/1166 (36.7%)。 来源:CC BY-NC 4.0 [@own_2_2024],使用 needle [@EMBL_tools_2022] 制作。
到目前为止,我们只讨论了双序列比对,其中两条序列被完整比对;这些被称为**全局比对 (global alignment)**。
局部比对¶
我们在第一章中看到许多蛋白质由结构域组成。 因此,某些序列可能在其全长范围内不相关,仅在对应于结构域的部分序列中共享相似性。 当比较此类蛋白质时,进行**局部比对 (local alignment)** 更为合适。 局部比对也是识别功能位点的良好工具,从中可以推导出序列模式和模体。
局部比对的目标是找到两条输入序列的最佳子序列,使其在给定比对参数下产生最大比对得分。 与全局比对一样,存在高效的算法来解决这个任务。 Smith-Waterman 算法可以在与输入序列长度的平方成正比的时间内解决这个任务,就像用于全局比对的 Needleman-Wunsch 算法一样。

使用不同参数对相同两条序列的比对:一次使用全局比对程序 needle [@EMBL_tools_2022],一次使用局部比对程序 water。使用相同的比对参数:DNAfull 矩阵,空位开启 -10,空位延伸 -0.5。全局一致性为 20/71 (28.2%),局部一致性为 12/12 (100.0%)。来源:CC BY-NC 4.0 [@own_2_2024]。
序列数据库搜索¶
在第一章中,我们学习了不同的序列数据库。 我们经常想在这些数据库中搜索新序列,例如了解哪些其他生物具有同源序列。 两条高度相似的序列也可能具有相同的功能。 这种关系被用于序列的功能注释,其中数据库搜索是一个重要步骤。
注意 2.3:随机相似性
当所有核苷酸以相同频率随机出现时,长度为 x 的每个序列出现的预期频率为 ¼x,例如长度为 3 的序列频率为 1/64,长度为 10 的序列频率约为百万分之一。 由于如今的数据库非常大——可以包含数百万条序列——这变得非常重要。 由于数据量巨大,某些相似性可能仅仅是偶然出现的,特别是当我们感兴趣的序列较短时。 因此,统计方法被开发出来,用于估计观察到的比对是否仅仅由于偶然而发生(见下文)。
数据库搜索与双序列比对¶
在数据库中搜索序列时也使用双序列比对。 在此任务中,我们有一条查询序列 (query),想要在数据库中找到相似的序列;这些相似序列称为__目标序列 (subjects)__ 或__命中 (hits)__。 虽然前面讨论的算法在比对两条序列时相对较快,但如果对数据库中所有潜在目标序列与查询序列逐一进行双序列比对,总体时间仍然太长。 因此我们需要更高效的算法。
注意 2.4:启发式算法
上一节描述的 Needleman-Wunsch 和 Smith-Waterman 算法保证能为给定序列和参数找到最佳得分的比对。 相比之下,启发式算法 (heuristic algorithm) 采用一些经验法则,通常能产生良好结果,并使算法运行更快。 然而,这种方法不再保证找到最优得分。
BLAST¶
Basic Local Alignment Search Tool (BLAST) 是一种启发式方法,用于发现蛋白质或核苷酸序列之间的局部相似区域。 该程序将核苷酸或蛋白质序列与数据库中的序列进行比较,并计算匹配的统计显著性。 BLAST 的独立版本和网络版本均可从美国国家生物技术信息中心 (NCBI) 获取。
算法¶
BLAST 的出发点是两条序列共有的"词 (word)"的集合,其中__词__是序列中固定长度的一部分。 对于蛋白质 BLAST,默认词长为 5,对于核苷酸 BLAST,默认词长为 11。 为了找到这些共有词,首先构建查询词的查找表,其中也列出了邻域词 (neighborhood words)。 邻域词__是所有与查询词具有高比对得分的词。 然后,BLAST 扫描数据库寻找词匹配。 对于蛋白质 BLAST,必须在 40 个残基内找到两个匹配,BLAST 才会将其视为__初始命中 (initial hit)。 请注意,对于核苷酸,初始命中的发现方式更简单:只需找到一个精确匹配,即不考虑邻域。
找到初始命中后,BLAST 将其扩展为局部比对。 随着这种__扩展__的进行,比对得分增加或减少。 当比对得分降至设定水平以下时,扩展停止。 这防止了比对延伸到查询序列和命中序列之间相似性很低的区域。 如果获得的比对得分超过某个阈值,它将被纳入最终的 BLAST 结果。 因此,BLAST 是一种启发式算法(注意 2.4),但其严谨的过程在运行时间和准确性之间提供了合理的权衡。
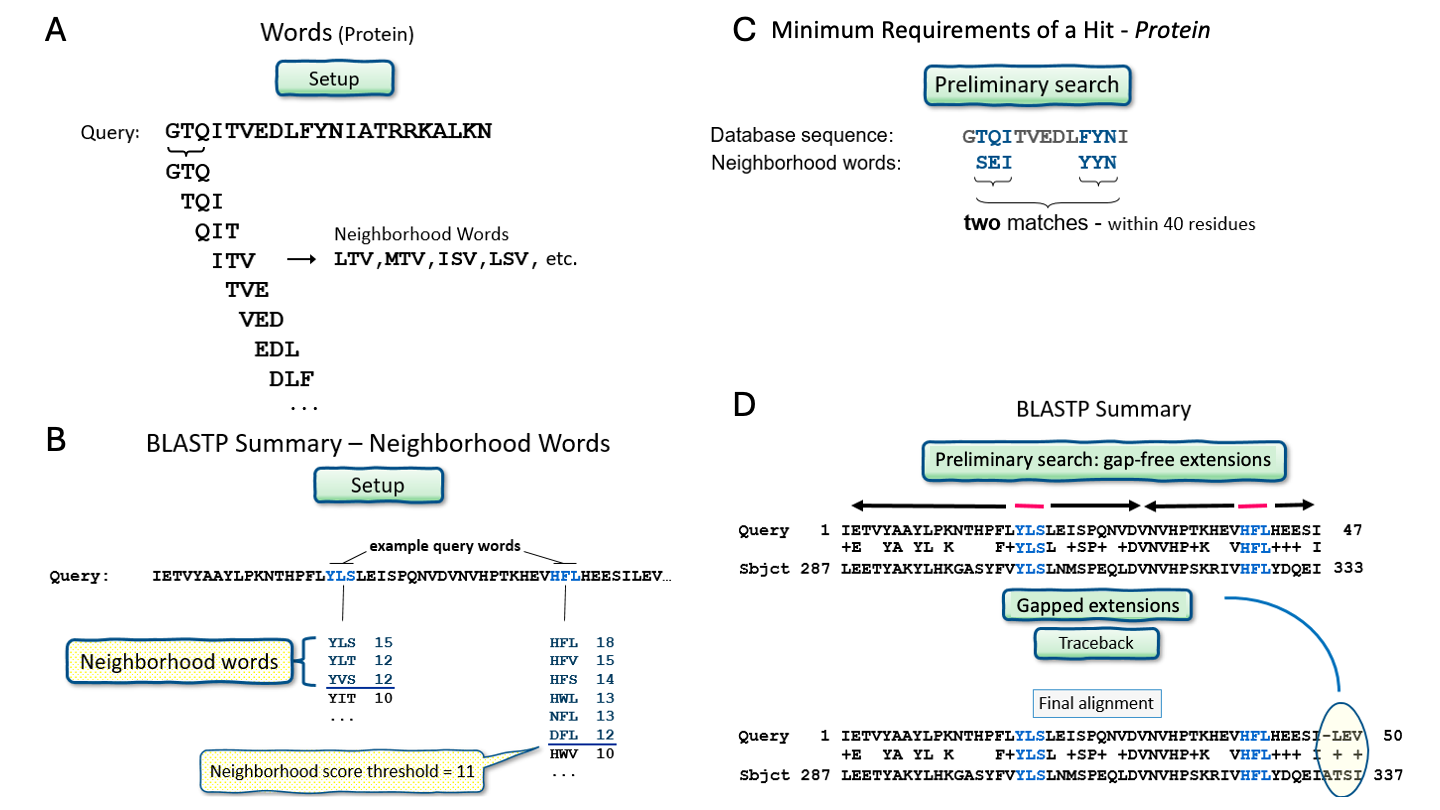
BLAST 算法概览。来源:CC0 1.0 [@blast_2022]。
BLAST 输出¶
BLAST 输出包含有关找到的命中、比对和分类的大量信息。
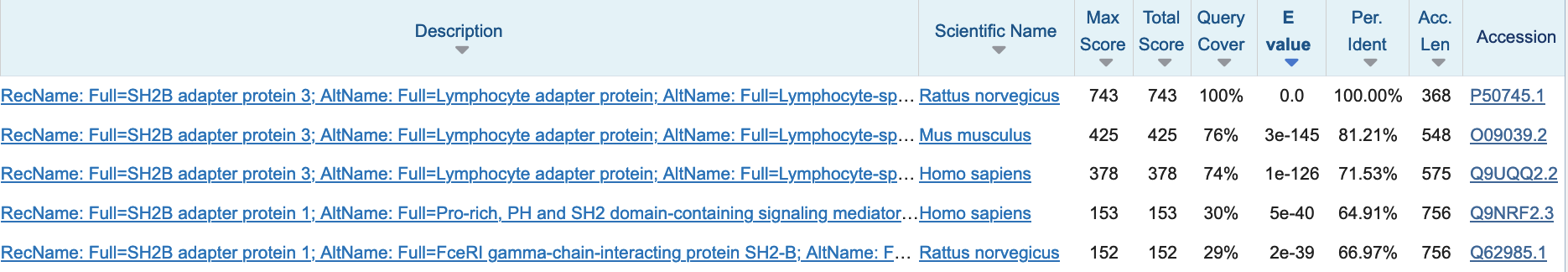
在 Swiss-Prot 2024_02 发布数据库中搜索大鼠蛋白质 P50745 的前 5 个 BLAST 命中。来源:[@blast_2009]。
注意 2.5:E 值
一个重要的输出统计量是期望值 (E-value),即在数据库中预期偶然出现具有观察得分或更高得分的 BLAST 命中数量。 请注意,根据此定义,E 值取决于数据库大小。 由于在较大的数据库中更可能偶然发现匹配,因此同一命中在较大数据库中的 E 值比较小数据库中更高。 因此,为了找到尽可能多的良好命中,应使用包含所有感兴趣序列的最小特定数据库。 例如,如果你只对植物感兴趣,则将搜索限制为仅植物序列。 在实践环节中,你将了解如何在在线 BLAST 界面中进行此操作。
BLAST 输出按 E 值递增排序。这可能导致非常小的数字,BLAST 输出使用科学计数法列出这些数字,例如 3e-145 表示 \(3 \times 10^{-145}\)。 因此,以下列出的命中不太可能是随机的,因为偶然出现的预期命中数量非常低。 如果比对不是偶然发生的,那么可能是由于两条序列之间存在生物学上有意义的关系。 然而,很难为生物学上有意义的命中定义一个明确的 E 值截断值。 常用的截断值为 1e-5 或 1e-10。
请注意,仅凭 E 值不能推断同源性;还需要考虑覆盖率和一致性百分比。 例如,在以下输出中,所有命中都具有非常低的 E 值: 第一个命中是序列本身;然后是在小鼠和人类中具有高一致性和高覆盖率的命中,这些可能是同源序列。 第 4 和第 5 个命中是局部的,因为查询覆盖率约为 30%,这些序列可能与查询蛋白质共享一个同源结构域。
BLAST 类型¶
存在不同类型的 BLAST,用于在各自数据库中搜索核苷酸或蛋白质: blastn 在核苷酸数据库中搜索核苷酸序列,blastp 在蛋白质数据库中搜索蛋白质序列。 此外,查询序列和/或数据库也可以在所有六个阅读框中进行翻译,以允许更多类型的比较。
不同类型的 BLAST 适用于这些不同类型的比较,这些翻译是自动完成的。
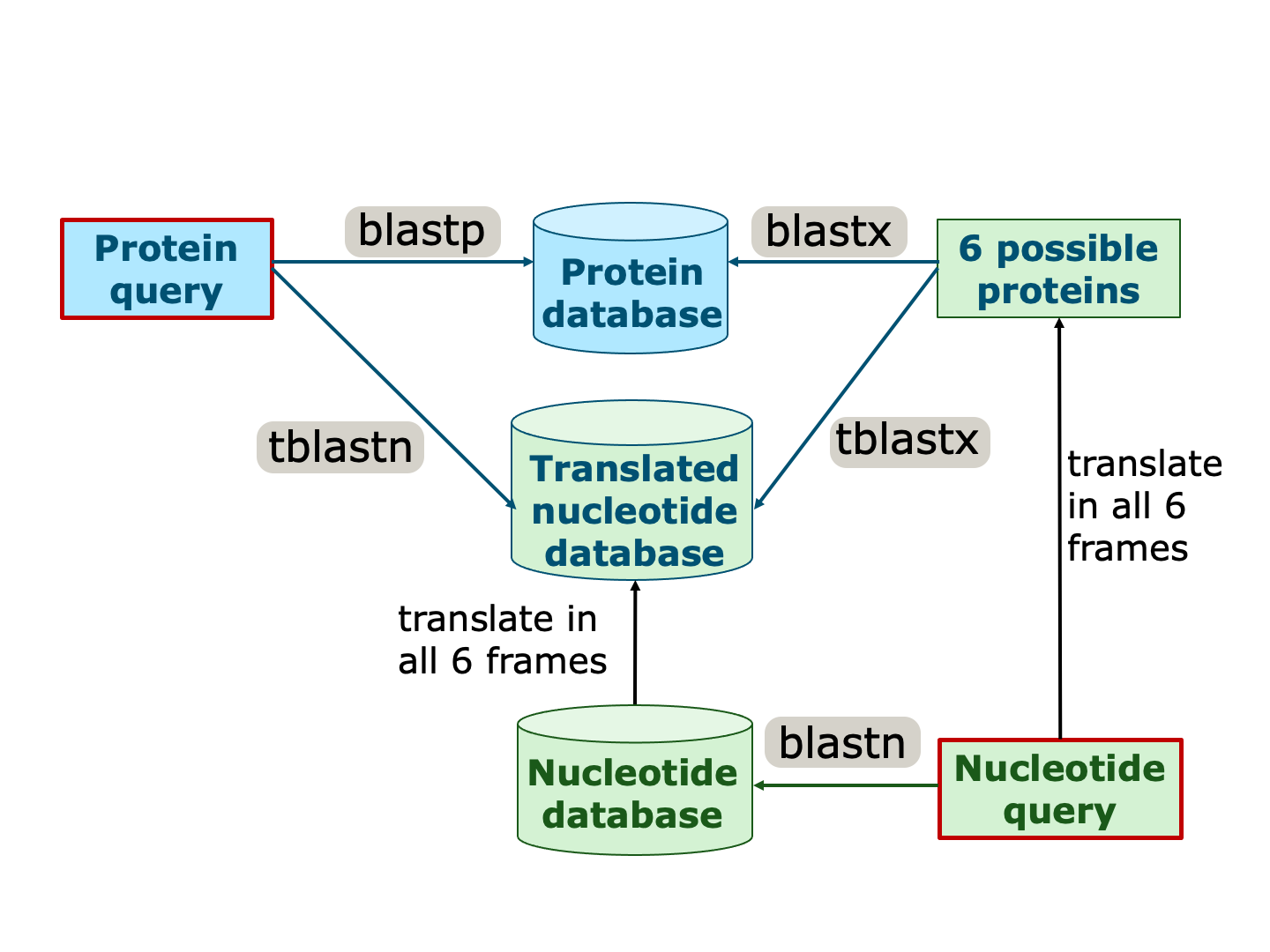
用于比较不同数据类型的不同 BLAST 类型。输入查询以红色框标记。蓝色背景的序列/数据库来自蛋白质序列,绿色背景的序列/数据库来自核苷酸序列。请注意,后者中的一些会自动翻译为所有可能的蛋白质(以蓝色字体表示)。来源:CC BY-NC 4.0 [@own_2_2024]。
PCR 引物设计¶
序列比较的一个特殊用途是为聚合酶链式反应 (PCR,见框 2.6) 设计__引物 (primers)__。 生物医学应用中的许多实验技术依赖 PCR 来扩增特定的 DNA 片段。 实例包括病原体检测、分析遗传变异、定点诱变、de novo 蛋白质合成和研究基因表达模式。 扩增哪些 DNA 片段主要由使用哪些 PCR __引物__决定。 为了设计能成功扩增目标 DNA 的引物,需要结合多个计算步骤。 本节重点介绍其中一些生物信息学方面的考虑。
框 2.6:聚合酶链式反应 (PCR)
聚合酶链式反应由 Kary B. Mullis 于 1983 年发明,于 1985 年在一项关于镰刀形细胞贫血症的研究中首次发表 [@saiki1985enzymatic]。在其发现十年后,PCR 的众多生物医学应用使其发明者获得了 1993 年诺贝尔奖(与 Michael Smith 因其定点诱变工作共享)。
作为一种扩增 DNA 的方法,PCR 依赖于 DNA 聚合酶催化的 DNA 复制这一自然过程来复制 DNA(见第一章)。 该反应使用所谓的引物来选择要扩增的 DNA 区域,并通过温度循环方案在每个循环中将反应产物的数量翻倍。 PCR 引物是相对较短的单链 DNA 片段,用于"引发 (prime)"聚合酶:它们决定 DNA 复制应从哪里开始。 引物总是成对出现:通过在所需 DNA 区域的两端和两条链上分别使用正向引物和反向引物,确保可以从一个原始 DNA 区域产生拷贝。
在反应过程中,通常交替进行三个不同的温度阶段: 1. 变性阶段 (~95°C) 将双链 DNA 解开为单链 DNA。 2. 退火阶段 (~55°C) 允许引物与其互补 DNA 结合,形成一个小区域的双链 DNA。 3. 延伸阶段 (~72°C) 允许聚合酶延伸双链区域,产生两个完整的原始材料双链拷贝。
重复此过程不断使拷贝数量翻倍,因此被称为链式反应。 在发明用于生物医学应用的 PCR 反应中一个关键的发现是使用能够承受变性阶段高温的聚合酶。 第一个耐热聚合酶是从生活在温泉中的一种细菌中提取的:Thermus aquaticus(因此,它以该物种命名为 Taq 聚合酶)。
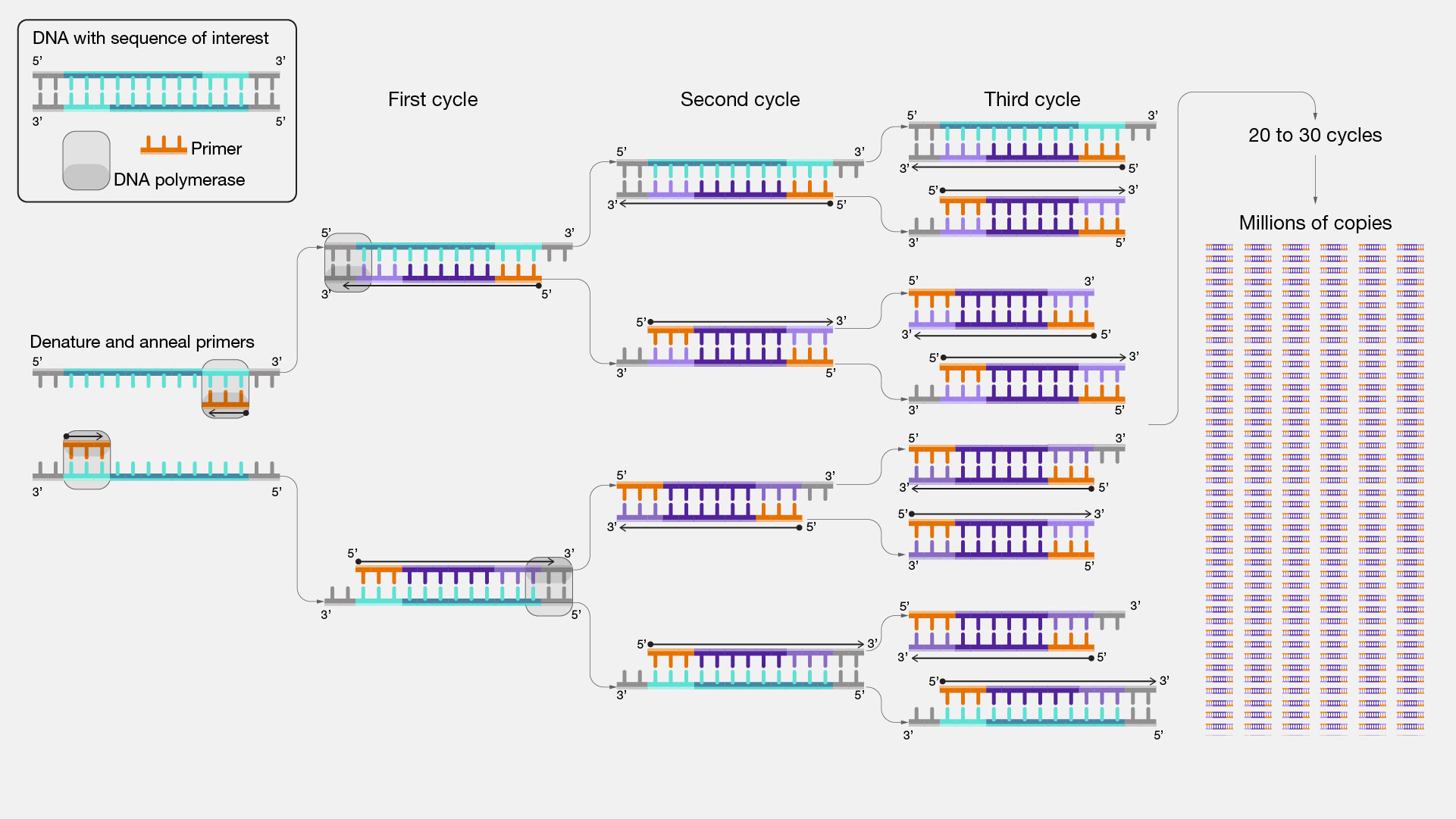
聚合酶链式反应使用引物选择所需的 DNA 区域,每个循环使反应产物翻倍。第一个循环后预期有 \(2^1=2\) 个拷贝,第二个循环后 \(2^2=4\) 个,第三个循环后 \(2^3=8\) 个... 来源:[@PCR_NHGR]。
PCR 引物通常需要满足多项要求才能产生成功的 PCR 产物:它们必须在生化上可行(即在正确的温度下变性、退火和延伸),它们必须具有特异性(仅扩增目标区域),它们应产生合理大小的产物(约 500-1000 个核苷酸,取决于具体应用),并且它们作为单链 DNA 应该是稳定的。 这些要求的结合通常允许引物长度为约 18-30 个核苷酸。 为了帮助快速设计可能成功的引物,Primer-BLAST 或 Primer3+ 等工具会自动检查上述提到的大多数要求。 例如,Primer-BLAST 允许用户上传需要扩增的 DNA 序列,并可以配置为找到特定大小的引物产物。 此外,通过在选择的数据库上使用 BLAST 检查可能的脱靶扩增(以确保特异性),还可以配置多个所需温度。
近似 PCR 变性温度 \(T_m\)
溶液中约一半 DNA 链处于变性状态的温度称为_熔解温度_ \(T_m\),是引物设计中的重要参数。 精确的熔解温度取决于 DNA 片段的确切长度和核苷酸组成,但对于短序列存在一个有用的近似公式。 该近似公式可用于快速检查和预测。
对于短于 14 个核苷酸的引物,熔解温度可以用以下公式近似计算:
\(T_m \approx 2 \times (A + T) + 4 \times (G + C)\)
其中 A、C、G 和 T 分别是引物中各核苷酸的数量。
多序列比较¶
多序列比对 (Multiple sequence alignment)¶
序列搜索的一个直接观察结果是,一条查询序列通常与多条序列相似。 这可以引发关于进化(这些序列来自哪里?)、功能(为什么某些序列彼此之间比与其他序列更相似?)或结构(这些序列的所有部分是否同样相似/不相似?)的研究问题。 使用双序列比对策略将所有这些序列相互比较会很快导致大量的比较,并且难以解释。 相反,当我们想要比较 3 条或更多序列时,我们使用**多序列比对 (Multiple sequence alignment, MSA)**。
执行多序列比对的目的是在可能具有不同长度的多条序列中识别匹配的残基(DNA、RNA 或氨基酸)。 得到的比对可以看作一个方阵:行代表我们开始的序列,列代表序列之间的同源残基,条目是残基或空位。
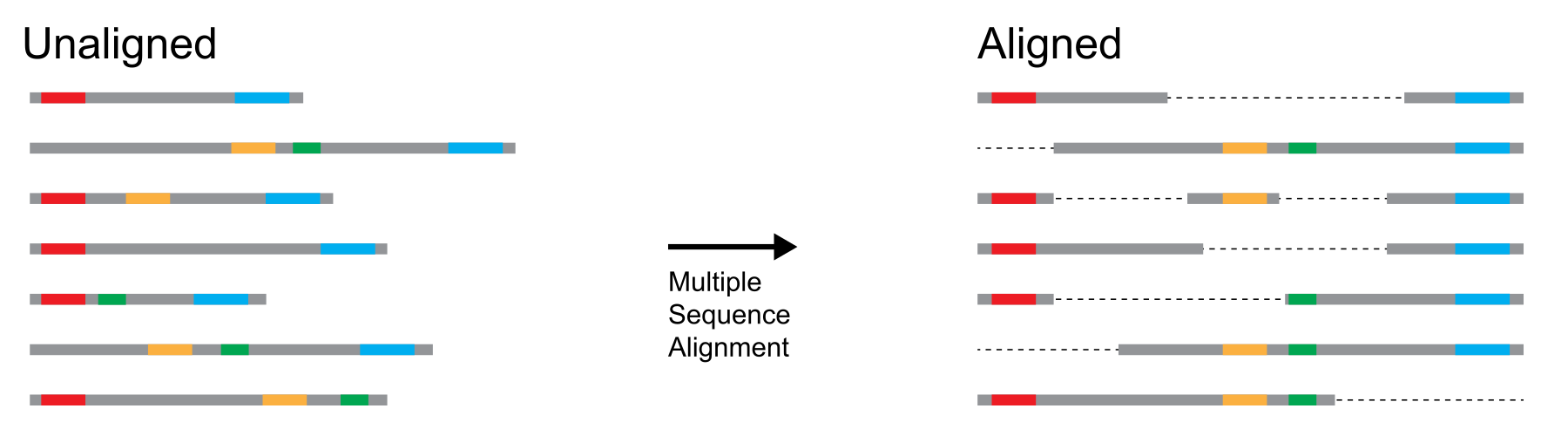
多序列比对的概念图解。彩色圆点表示相似的序列元素,在右侧的多序列图中这些元素在垂直列中对齐。来源:CC BY-NC 4.0 [@own_2_2024]。
与双序列比对不同,对于一组序列和评分参数,计算最佳多序列比对在计算上是不可行的。 相反,存在各种用于创建多序列比对的启发式算法(注意 2.4)。 这里我们将介绍许多工具采用的两种主要概念:渐进比对和迭代优化。
渐进比对¶
渐进比对使用所谓的__指导树 (guide tree)__(框 2.2)来构建比对。 指导树是所有要比对序列之间相似性的粗略表示。 渐进比对使用指导树选择两条最相似的序列,并通过全局比对策略将这两条序列比对来初始化多序列比对。 随后,指导树用于确定序列添加到比对中的顺序。 可以这样理解渐进比对:它创建越来越大的序列"块",其中块始终被视为一个单位(例如,引入空位将对块中的所有序列发生)。 通过遍历指导树,这种比对策略_逐步推进_到最终结果,因此得名_渐进比对_。
框 2.2:构建指导树
渐进比对策略使用的指导树通常由聚类算法创建,该算法以所有序列之间的两两距离作为输入(见第三章)。 获取这些两两距离可以通过例如局部比对得分来完成,但另一种常见方法是计算两条序列中都存在的长度为 \(k\) 的子序列(也称为 \(k\)-mers)的数量。 这种基于 \(k\)-mer 的策略的缺点是它提供了粗略的距离度量(因此不太准确),好处是非常快。
一旦使用渐进策略创建了多序列比对,就可以基于这个第一个多序列比对重新计算指导树,并根据更新后的指导树计算第二个多序列比对。 从理论上讲,重新计算指导树可以重复很多次(例如,直到不再变化为止),实际上只需重新计算一次就足够了。 常用的多序列比对程序 mafft 在 FFT-NS-2 算法中实现了指导树的重新计算。
迭代优化¶
渐进比对策略的一个潜在缺点是,某些中间块可能代表次优比对。 例如,当在渐进方法的早期阶段引入空位时,它永远不会从比对中移除。 识别并可能改善此类情况通常被称为__迭代优化 (iterative refinement)__,通常在由渐进策略创建的多序列比对上进行。
迭代优化以一个多序列比对、一个多序列比对的评分函数和一个重新排列多序列比对的函数作为输入。 它通过重新排列多序列比对产生一个_优化的_多序列比对,只有在得分增加时才保留新的多序列比对。 这个过程通常重复进行,直到得分不再增加(或进行固定次数的迭代)。
由于迭代优化方法通常从渐进比对开始并改善其得分,实现迭代优化策略的程序(例如 mafft 中的 FFT-NS-i 方法)通常比基于渐进比对的程序(例如 mafft 中的 FFT-NS-2 方法和 Clustal 程序)表现更好,但也需要更多时间 [@katoh_mafft_2014]。
框 2.3:多序列比对的评分与重新排列
对于迭代优化,存在各种评分和重新排列策略。这里我们概述一种常见的方法:加权配对和 (weighted sum-of-pairs) 评分函数和分块重新排列策略。
加权配对和评分:配对和方法计算并求和所有可能的双序列比对得分。加权版本包括为每对添加特定的权重因子,其中权重由序列之间的系统发育关系决定。
分块重新排列:按照指导树,多序列比对沿着树的每个分支被划分为两个子比对(或块)。然后对每对块进行重新比对,但只有当重新比对的块的得分增加时才保留新的比对。
模体 (Motifs)¶
在了解了如何获得多序列比对之后,我们现在关注几种解读方式。 一种可能的解读是识别(并搜索)常见的序列模式。 常用术语__模体 (motif)__指常见的序列模式,我们从现在开始使用这个术语。 模体可以通过总结多序列比对的_列_来发现,试图描述所有序列中常见的残基。
注意 2.5:多序列比对与模体
由于模体基于多序列比对,可能会有人想互换使用这两个术语。一个关键区别是模体始终代表一个常见的模式,而多序列比对也可能包含保守性/相似性较低的区域。此外,一个多序列比对可以包含多个模体。
模体最简单的表示是__一致序列 (consensus sequence)__,其中多序列比对的每一列由最频繁出现的残基表示(即多数一致序列)。 一致序列的缺点是它不能表示模体中存在的任何变异。
一致序列的一种扩展是可以表示模体中一些变异的__模式字符串 (pattern string)。 在模式字符串中,明确的位置由单个字母表示,并表示变异的特殊语法: 多序列比对中具有多个字符的位置用方括号内的多个字符表示。 例如,包含模式 [AG] 的模式字符串表示模体中的一个位置可以是 A 或 G。 因此,模式字符串从正则表达式中汲取灵感。 存在各种类型的模式字符串;例如,Prosite 数据库中使用的 PROSITE __REF 字符串包含用于表示模体中残基无关位置的语法(用 * 标记)。 模式字符串能够表示模体中的一些变异,但不能表达特定变异出现的可能性有多大(在 [AG] 的例子中,A 和 G 出现的可能性相同)。
为了表达特定残基在特定位置出现的可能性,可以使用__位置特异性评分矩阵 (Position Specific Scoring Matrix, PSSM)__。 每一行代表多序列比对中可能的字符之一,每一列代表多序列比对中的一列,数字指示在特定位置观察到特定字符的概率。 因此,每列的概率之和为 1。 例如:一个 DNA PSSM 将有四行,代表核苷酸 A、C、G 或 T。 条目代表在特定位置观察到特定残基的概率。 因此,PSSM 中所有列的总和必须为 1。 由于 PSSM 包含概率,因此可以相对简单地计算未知序列与现有 PSSM 的匹配程度:假设位置之间独立,只需将新序列中字符的观测概率相乘即可。
最后,序列标志图 (sequence logos) 是比对的图形表示。 序列标志图中的每个位置代表多序列比对中的一个位置。 标志图在该位置的总高度指示该列包含的_信息量_,即明确的位置具有高信息含量,而字符频率相等的位置具有低信息含量。 此外,字符按其在各自位置上被观测到的概率成比例缩放。
![]()
保守模体的各种表示方式的概念图解。A:5 条序列和 7 个位置的多序列比对 (MSA)。B:一致序列。C:模式字符串。D:位置特异性评分矩阵 (PSSM)。E:序列标志图。来源:CC BY-NC 4.0 [@own_2_2024]。
概隐马尔可夫模型 (profile hidden Markov models, pHMMs)¶
前面关于多序列比对和模体的章节解释了如何总结和使用相似序列集合的一些基础知识。 在本节中,我们强调一种利用多序列比对中的信息进行序列搜索和比较的强大方法:概隐马尔可夫模型 (profile hidden Markov models, pHMMs)。 一般隐马尔可夫模型的一些基础知识已在第一章中涵盖;这里我们介绍对 HMM 一般概念的一些简单改编如何解锁强大的序列搜索方法。
我们可以将概隐马尔可夫模型视为位置特异性评分矩阵的扩展。 与 PSSM 一样,pHMM 包含在多序列比对中特定位置观察到某些字符的概率。 然而,与 PSSM 不同的是,HMM 还可以表示插入和缺失,即在一个位置插入的可能性可能比在另一个位置插入的可能性更大。 为此,隐马尔可夫模型包含_隐状态_匹配/插入/缺失以及隐状态之间的_转移概率_。 此外,_发射概率_代表匹配状态和插入状态中不同字符的各自概率。 简单概 HMM 的图形表示可以参见下图。 与 PSSM 一样,可以为与现有 HMM 匹配的新序列计算概率得分。 存在用于处理 pHMM 的高效算法,并已在例如 HMMer 套件中实现。
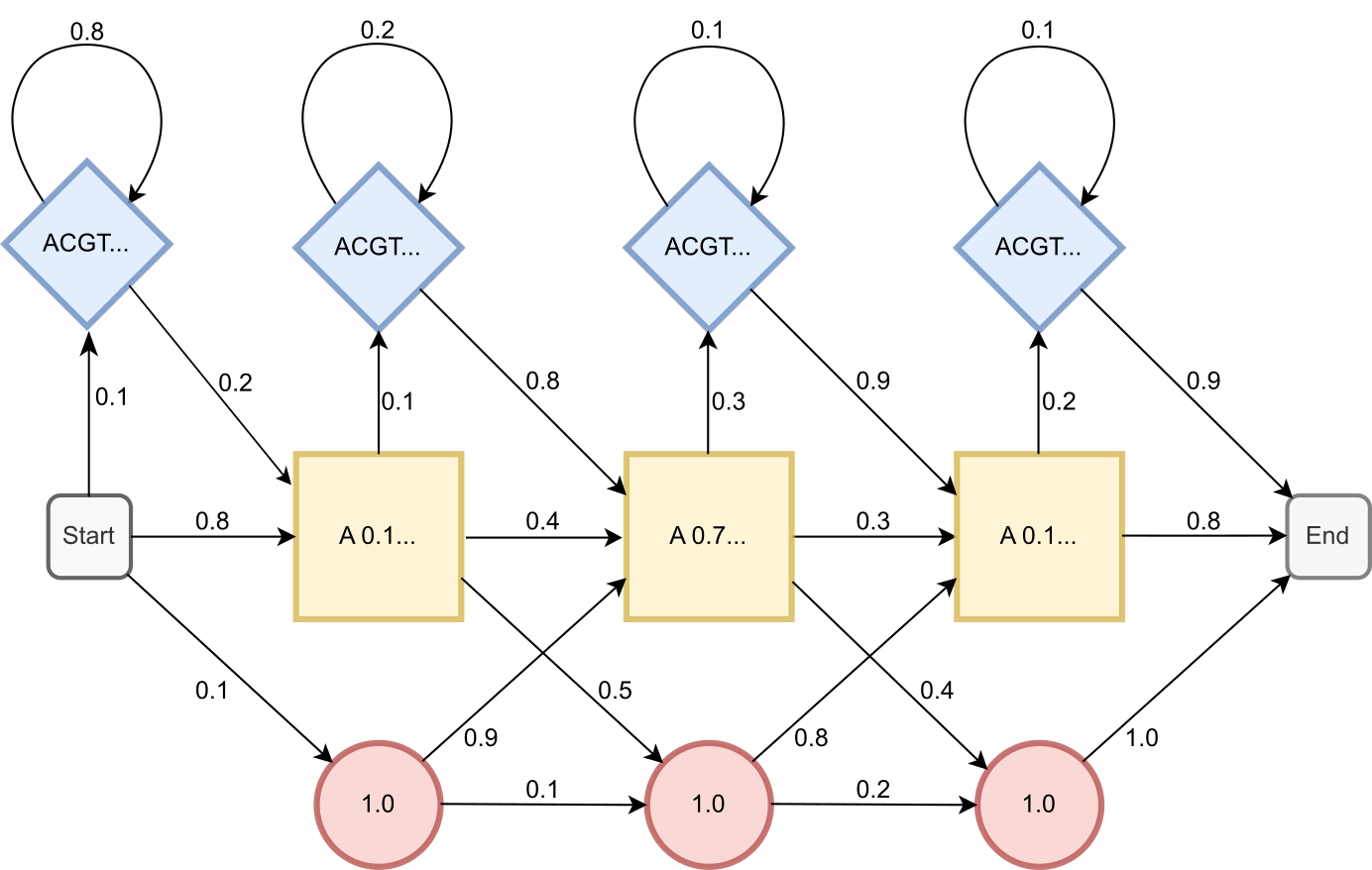
包含所有模型概率的简单 DNA 概 HMM 示意图。模型由三种类型的隐状态组成:匹配(黄色方块)、缺失(红色圆形)和插入(蓝色菱形)。发射概率标示在隐状态内部,隐状态之间的转移概率标示在箭头旁边。来源:CC BY-NC 4.0 [@own_2_2024]。
框 2.4:计算序列的概率
原则上,上图中的信息足以计算序列属于该 pHMM 的概率。 例如,由匹配状态表示的序列 GAT 的概率为 \((0.8 * 0.6) * (0.4 * 0.7) * (0.3 * 0.4) * 0.8 = 0.013\)。 我们通过将所有相关的转移和发射概率相乘得到这个数字。 请注意,隐状态通常是未知的;因此,确定相关概率比这个简单示例更复杂。 存在用于确定通过 HMM 图最优路径的高效算法,但超出了本书的范围。 另外,我们不期望你能手动进行这些计算。
使用多序列比对进行序列搜索
将多序列比对转换为概率集合(例如 PSSM 或 pHMM)的能力使得计算新序列_属于_该多序列比对的概率成为可能。 这种技术通常提供比基于双序列比对(例如 BLAST)的搜索更敏感的方法。 在实践中,这意味着可以在更大的进化距离上识别匹配序列。 实现此方法某些版本的工具包括 psiBLAST(使用 PSSM)和各种 HMMer 工具(均使用 pHMM)。
框 2.5:数据库中的 pHMM
基于保守/共现区域将生物序列分组,然后利用这种分组进行序列搜索的能力,在广泛的生物序列数据库中被利用。 其中一些数据库已在第一章中介绍;这里我们以 Pfam 为例,简要概述 HMM 如何被纳入这些资源中。 Pfam 数据库中的所有条目都由概 HMM 表示。 这些条目被分为六类之一:家族 (family)、结构域 (domain)、重复 (repeat)、保守位点 (conserved site)、卷曲螺旋 (coiled coil) 或无序 (disordered)。 这六类之间的主要区别是匹配序列的长度:家族 Pfam HMM 预期在蛋白质序列的全长范围内匹配,_保守位点_通常只是蛋白质中的一小区域。 因此,多个 Pfam HMM 可以匹配给定的蛋白质序列。在给定序列上匹配的 Pfam HMM 的组合可用于给出序列中已知元素的细粒度描述。
实践练习¶
本实践环节包含问题和练习,帮助你处理第 2 章的学习材料。 有两个有指导的实践环节,分别在周三和周四。 在第一个实践日,你应该完成本指南的大约一半。 因此,你应该在第一天接近完成练习 4。 利用时间指示确保你不会困在一个练习上。 这些实践练习为你提供项目前的最佳准备。 确保你现在培养实践技能,以便在项目期间应用它们。
注意,答案将在实践后发布!
Vps36 蛋白质序列 BLAST,30 分钟
寻找蛋白质的同源物是生物学中的常见任务,因为同源物的存在可以告诉我们蛋白质的功能以及在哪些其他物种中可以找到。因此,我们首先练习使用 BLAST 寻找同源物。我们想要找到酵母蛋白质 Vps36 的同源物,即液泡蛋白分选相关蛋白 36 (Vacuolar protein-sorting-associated protein 36)。为此,我们在不同数据库中搜索并比较结果。 1. 从 NCBI 以 FASTA 格式获取酿酒酵母 (S. cerevisiae) 的 Vps36p 蛋白质序列 (NP_013521.3)。(注意:你可以将其下载到文件中,或保持标签页打开并使用复制/粘贴)。FASTA 格式是什么样的?即序列和序列的附加信息(名称)是如何存储的? 2. 我们希望使用美国国家生物技术信息中心 (NCBI) BLAST 程序在 SwissProt 数据库中识别相似蛋白质。前往 NCBI 网站。在网页右侧,你可以找到 BLAST 的直接链接。接下来,你需要决定哪种搜索策略适合以蛋白质序列为查询在蛋白质数据库 (SwissProt) 中搜索(如果不确定,请查看上文关于 BLAST 类型的内容)。点击适当的搜索策略。将序列输入查询框,选择 UniProtKB/SwissProt 作为要搜索的数据库,并确保选择了正确的算法。最后,点击 "BLAST" 按钮执行搜索。先在 SwissProt 中搜索可能有什么优势和劣势? 3. 查看搜索结果页面。你可以在哪里找到关于查询的信息、数据库命中的概览、这些命中沿查询序列的分布,以及查询与每个数据库序列之间的比对? 4. 你在哪些生物体中找到相似序列?你认为哪些命中是同源物? 5. E 值的解释是什么?E 值为 0.0 意味着什么?注意:我们仍然需要 SwissProt 中的结果。保存 URL,以便稍后检索,或者保持这些结果打开,在新浏览器窗口或标签页中执行新搜索。 6. 接下来,我们想要找到更多同源序列;因此在 ClusteredNR 数据库中搜索。你找到多少个命中?查看最后一个命中,你认为该数据库中的所有相似序列都已被找到吗? 7. 执行与 (6) 相同的搜索,但允许找到更多的命中(将算法参数下的 Max target sequences 更改为最大可用值)。你找到多少个命中?你会将它们全部视为酵母 Vps36 的同源物吗? 8. 返回 SwissProt 结果,查看第一个不是查询序列本身的命中。将 BLAST 结果(得分、查询覆盖率、E 值、一致性百分比、比对长度)与 ClusteredNR 中发现的同一物种的命中进行比较。你观察到了什么?
更多 BLAST 搜索,60 分钟
BLAST 不仅可以使用 blastp 在蛋白质数据库中搜索蛋白质序列,还提供不同的 BLAST 类型,允许在不同类型的数据库中搜索或使用替代搜索策略,以及将搜索限制在特定物种。 在这里,我们通过在真菌和其他生物体中搜索酵母 Vps36p 的同源物来探索这些策略。 1. 接下来,我们想知道这种蛋白质在新型隐球菌 (Cryptococcus neoformans) 中是否存在同源物。最直接的 BLAST 搜索策略是什么?使用大型数据库 (ClusteredNR) 执行此 BLAST 搜索。你找到多少个命中?检查比对的长度、一致性百分比和 E 值。你观察到了什么,你得出了什么结论? 2. 如果使用"普通" blastp 搜索没有找到同源物,你可以使用哪些替代方法来仍然找到同源物(例如,在基因组序列中)?描述该 BLAST 类型中发生了什么。 3. 使用 tblastn 将 Vps36p 的蛋白质序列在新型隐球菌 (C. neoformans) 的核苷酸序列中搜索,指示你_仅_想搜索这一个物种而不是整个数据库(使用数据库:Core nucleotide database (core_nt))。检查搜索结果。你认为这些是好的命中吗?你能得出新型隐球菌中存在(或不存在)该基因同源物的结论吗? 4. 报告的一些命中是新型隐球菌 1 号染色体的一部分。更详细地检查这些命中。你的查询有多长,数据库中的序列有多长? 5. 考虑以下情况,你想更详细地研究这个命中。例如,你可以将你的蛋白质与这个数据库命中以及其他序列进行多序列比对。如果你从数据库中下载序列会发生什么?为什么这可能是一个问题,你如何解决它? 6. 酿酒酵母 (S. cerevisiae) 属于子囊菌门 (Ascomycota),而新型隐球菌 (C. neoformans) 属于担子菌门 (Basidiomycota)。Vps36p 在整个子囊菌门中高度保守,并且可能在该门之外也有同源物,正如你上面的搜索所示。我们现在想要更好地了解 Vps36p 在其他物种(子囊菌门之外)中可能的同源物。为此,在 refseq_protein 数据库中(排除子囊菌门)对 Vps36p 执行 blastp 搜索,将目标序列数设置为最大。查看最佳命中。关于查询覆盖率和一致性你能说什么? 7. 查看结果的分类报告(BLAST 结果页面顶部的链接)。你在哪些物种类群中找到命中? 8. 可能的 BLAST 策略来找到更远的同源物有哪些? 9. 尝试修改 (6) 中搜索的算法参数。为了找到更多同源物,继续使用 blastp 算法,只是尝试修改一个参数。你找到多少个命中? 10. 查看分类报告。你现在在哪些物种类群中找到命中? 11. 最后,我们想了解这些远缘相关蛋白质的相似程度。为此,我们将下载一些命中并执行多序列比对。生成一个包含 10 条序列的 multi-fasta 文件:前一个 BLAST 搜索的前 9 个命中和原始序列 (NP_013521.1)。(提示:你可以标记序列并通过点击 Download -> Fasta (complete sequences) 来保存它们;使用文本编辑器手动添加原始序列)。 12. 我们将使用 T-Coffee 套件中的 M-Coffee。该程序使用多个其他工具计算多个多序列比对,并将它们合并为一个最终比对。输出包括显示方法之间一致性的颜色编码。上传你的 multi-fasta 文件并使用默认参数运行。查看估计的比对。关于整体比对质量你能说什么?你可以在哪里找到高一致性和低一致性的区域? 13. 你是否认为这些序列在全长范围内同源?为什么/为什么不?
寻找远缘同源物,20 分钟
除了 BLAST,还存在其他同源搜索方法,其中一些对于发现远缘同源物特别有用。在这里,我们将使用其中一个工具 HMMER。 1. 我们首先使用 phmmer 进行搜索。phmmer 可以像 BLAST 一样使用,它在序列数据库中搜索序列。在内部,phmmer 使用 BLOSUM62 和空位从你的单条查询序列构建概模型。这样,它更_敏感_,即预期与 BLAST 相比能发现更多远缘同源物。使用 phmmer 在 SwissProt 数据库中搜索 Vps36p。你找到多少个命中?它们与练习 1 问题 2 的结果相比如何? 2. 接下来我们尝试使用基于 HMM 的工具 jackhmmer 寻找更多远缘同源物。阅读这里的第一段以了解此工具。该方法如何在后续迭代中找到更多命中? 3. 使用默认数据库运行 jackhmmer。你找到多少个命中,它们在分类中如何分布? 4. 使用顶部按钮启动第二次迭代。你现在获得多少个命中?将分类与上一次迭代进行比较。你观察到了什么?
PLT1 家族分析 - 第 1 部分,60 分钟
干细胞是未分化的细胞,可以分化为特化细胞,因此在胚胎发育和生长中至关重要。 在植物中,如拟南芥 (Arabidopsis thaliana),干细胞存在于根和芽的特定区域,从而为这些组织提供持续的特化细胞供应。 PLT1 是维持根干细胞所需的转录因子(Aida et al. 2004)。 在这里,我们将使用生物信息学方法分析 PLT1,以发现它是否属于更大的基因家族以及在拟南芥中存在哪些相关序列。
UniProt 是一个公开可用的蛋白质数据库,包含超过 200,000,000 个蛋白质条目的蛋白质序列和功能注释。 1. 查看 UniProt 网站。为什么 UniProtKB-TrEMBL 比 UniProtKB-Swiss-Prot 有更多条目? 2. 使用 UniProt 标识符 Q5YGP8 搜索拟南芥蛋白质 PLT1。PLT1 条目为你提供蛋白质条目的概览和一些功能信息。阅读 PLT1 的功能描述。这个描述是否与上述关于 PLT1 的信息相符?UniProt 是如何收集这些信息的? 3. PLT1 中存在哪些功能区域?它们位于序列的哪里?用于该信息的数据库是什么?(提示:功能区域可以在 Function -> Features 下找到)。 4. 在条目的末尾附近,你可以找到 PLT1 的实际蛋白质序列。你可以通过点击 Download 按钮以 FASTA 格式下载该序列。 5. Interpro 也提供蛋白质及其结构域的功能分析。前往 Interpro 网站并查找 PLT1 的条目。在 PLT1 中已识别出多少个蛋白质结构域,它们位于哪里? 6. 在 Interpro 中查找该结构域。已识别结构域的功能是什么?你可以找到关于 GO 条目和蛋白质结构的什么信息? 7. 在 Pfam 中查找该结构域并查看该结构域的 HMM 标志图。哪 3 个位置最保守,那里偏好哪些氨基酸? 8. 我们想要使用在线点阵图程序 Dotlet 分析该蛋白质中的重复。前往网站并将 PLT1 蛋白质序列添加为序列 1 和序列 2(我们想要进行自身比较)。要过滤一些低得分比对,你需要使用得分直方图下方的滑块。你可以在该蛋白质片段中找到多少个重复,这些重复位于蛋白质片段的哪些位置? 9. 如果将评分矩阵从 BLOSUM62 更改为 Identity 会发生什么? 10. 使用鼠标点击可能包含重复序列的区域。使用左右箭头键定位比对重复结构的起始位置。你可以识别哪些保守氨基酸?将重复家族的标志图与你发现的保守氨基酸进行比较。你观察到了什么? 11. 双序列比对可以识别保守区域。获取在 InterPro 中发现的 PLT1 的第一个和第二个 AP2 结构域的氨基酸序列,并使用 EBI 网站上可用的算法执行双序列比对。首先使用 Needleman-Wunsch 算法 (Needle) 执行全局比对。选择蛋白质比对并分别添加 PLT1 各蛋白质结构域的序列。两个结构域之间的整体一致性和相似性是多少?为什么这两个值不同? 12. 现在使用 Smith-Waterman 算法执行局部比对。你预期全局比对和局部比对之间会有很大差异吗?解释原因。
PLT1 家族分析 - 第 2 部分,寻找同源物,30 分钟
在分析了 PLT1 序列之后,我们想在拟南芥 (Arabidopsis thaliana) 中寻找 PLT1 的潜在同源物。 我们将使用 BLAST 在公开可用的数据库中识别具有足够高相似性得分的蛋白质序列,使其很可能是 PLT1 的同源物。 1. 返回 UniProt 数据库并在 UniProt 条目中点击 Tools -> BLAST。将目标数据库更改为 "UniProtKB Swiss-Prot" 并执行搜索("Run BLAST")。找到的"最佳"命中是什么(你如何定义"最佳"?)查看第二佳命中,那是哪条序列,在哪个生物体中发现的?报告 E 值、一致性、得分。这两个序列可能有什么关系? 2. 你通过该搜索找到多少个命中? 3. 你预期在 UniProtKB 数据库中找到比原始搜索更多还是更少的数据库命中吗?为什么?哪种数据库是在植物中识别 PLT1 同源物最有用的数据库?为什么? 4. 你如何影响在数据库中找到的命中数量? 5. 重复搜索,但只考虑 E 值为 1e-4 及最高 1,000 个可能匹配的命中。你找到多少个命中? 6. 现在我们想集中在拟南芥 (A. thaliana) 中的同源物上(在 popular organism 中点击 A. thaliana)。你在拟南芥中找到多少个命中? 7. 在输出页面的右侧,你可以找到比对的图形概览(比对部分以粗条显示)。你还可以点击这些来查看比对区域。如果你查看前十条序列,关于比对区域你观察到了什么,这暗示了什么?这是你从 BLAST 搜索中预期的吗?为什么? 8. 以 FASTA 格式保存前十个数据库命中。注意:UniProt 提供比对选项,这是获取所有感兴趣序列的最简单方法:标记它们 -> 点击 Align -> 点击 Align selected results -> 将窗口中的序列复制到文本文件中,注意复制完整的 10 条序列。
PLT1 家族分析 - 第 3 部分,保守性,30 分钟
接下来,我们想探索在上一个练习中识别的 PLT1 家族的保守性。 为此,我们使用多序列比对。 1. 使用 SwissProt 数据库的前十个命中(见练习 5 问题 8)使用 MAFFT 执行多序列比对。以 FASTA 格式下载此比对并保存在你以后能找到的地方。你将在第三章中重用此比对来构建系统发育树。 2. 哪些区域比对良好,哪些不好?你如何在多序列比对中轻松发现这些?该区域与先前识别的蛋白质结构域有什么关系? 3. 查看 MAFFT 中作为选项可用的迭代优化方法。你认为哪种策略适合你的数据集? 4. 运行你提出的策略并将其与之前的比对进行比较。你观察到了什么?再次检查第一次运行的结果页面。你能为你的观察找到解释吗? 5. 在比对查看程序中显示你的第一个多序列比对(问题 1)。在比对中定位第一个 AP2 结构域的起始位置(提示:查看 Pfam 标志图并尝试找到前 2 个高度保守的位置)。结构域从哪里开始?查看结构域的前 10 个位置,将比对中的保守性与 Pfam 标志图进行比较。你观察到了什么? 6. 接下来,我们尝试一个不同的比对工具:T-Coffee 套件中的 M-Coffee。该工具是否提供良好的全局比对?为什么/为什么不? 7. T-Coffee 套件还包括一个从比对中提取可靠区域的程序:Core/TCS 工具。我们想使用此工具从我们的比对中提取可靠列。使用底部的 "Send results" -> Core/TCS 下的按钮运行它,然后点击 "Submit"。一个仅包含比对良好列的 FASTA 文件可以在底部下载("fasta_aln file")。在 mview 中显示此文件。关于此比对的质量你能说什么?
细菌中的模体发现,20 分钟
细菌免疫系统 CRISPR/Cas 在 CRISPR(成簇规律间隔短回文重复序列)基因座中编码防御序列以靶向可移动遗传元件,其中防御序列位于重复序列之间。 在这里我们将使用模体发现来确定重复序列。 1. 访问 NCBI 的 RefSeq 数据库以获取嗜热链球菌 (Streptococcus thermophilus) 的基因组数据 (Accession NZ_LR822015.1)。提示:使用 Customize view 显示所有特征。 2. 转到第一个 repeat_region 特征。它在基因组的什么位置?获取该特征的序列(提示:点击 repeat_region,然后点击右下角的 Fasta)。 3. 使用 MEME 和 MAST 发现模体。前往 MEME suite 并点击 MEME。在 Input 下,从下拉菜单中选择 "Type in sequences" 以粘贴你的 FASTA 序列。在 "How do you expect motif sites to be distributed in sequences?" 下选择正确的选项,并选择发现一个模体。 4. 运行搜索后,获取 MAST HTML 输出。你发现了什么模体,它在序列中出现多少次?将模体与 RefSeq 中注释的重复进行比较。你观察到了什么?
致病疫霉效应基因 Avr1 的引物设计,30 分钟
致病疫霉 (Phytophthora infestans) 是番茄和马铃薯晚疫病的病原体。 马铃薯晚疫病在欧洲和北美具有重大的历史影响,因为它导致了 19 世纪中叶爱尔兰的大饥荒,其中一百万爱尔兰居民死亡,另一百万移民到美国。致病疫霉与许多其他植物病原体一样,利用所谓的效应蛋白在易感植物宿主中建立自身。致病疫霉中的 Avr1 基因编码效应蛋白 Avr1,它有助于在易感马铃薯植株中的毒力,但在某些抗性马铃薯品种中被植物免疫系统识别。因此,为了避免植物免疫系统的识别,一些致病疫霉分离株丢失了 Avr1 基因。在这里,我们将通过为其序列 (XM_002896847.1) 设计引物来寻找 Avr1 的存在。最近,一位农民在瓦赫宁根附近的不同田间收集了不同的致病疫霉分离株,农民想知道这些分离株是否包含 Avr1。在这里,我们的目标是设计可以用来检测致病疫霉中 Avr1 基因是否存在的引物。 1. 为了帮助你设计"合适的"引物(记住设计引物时哪些特性很重要),我们将使用 PrimerBLAST。PrimerBLAST 结合了 Primer3(一个为给定目标序列设计引物的程序)和 BLAST(确定引物序列是否具有特异性)。 2. 查看预期产物大小的设置(默认值设置为 70-1000nt)。这是什么意思? 3. 我们要确保设计的引物尽可能具有特异性。因此,我们想避免设计的引物可以匹配基因组中目标区域以外的任何其他区域,在这种情况下是 Avr1。因此,我们可以指示 PrimerBLAST 将对致病疫霉基因组序列检查特异性。为此,选择数据库 ('Refseq representative genomes') 并在 'organism' 字段中输入 Phytophthora infestans。另一种可能性是使用 'non-redundant' (nr) 数据库。你能想象为什么在选择 nr 数据库时识别特异性引物序列可能有问题吗? 4. 在 PrimerBLAST 的搜索字段中输入 Avr1 的登录号,并使用问题 3 中定义的选项(数据库和物种)运行 PrimerBLAST。PrimerBLAST 将识别你的 Avr1 与数据库中的现有序列匹配,这可能干扰特异性引物的识别。为确保 PrimerBLAST 考虑到这一点,选择数据库序列,然后点击提交按钮继续。 5. 查看 PrimerBLAST 的结果。顶部,你将找到关于提交序列(其长度)的摘要,以及关于 PrimerBLAST 是否能够识别特异性引物的消息。下方,你将找到引物对沿序列分布的图形概览,以及每个引物对的详细信息(例如 GC 含量、Tm、长度和产物长度)。哪对引物最好,为什么(显然所有引物都满足质量标准)? 6. 如果将鼠标放在图形概览中的引物对上,你可以将引物和产物的序列保存为 FASTA 格式文件。此外,你还可以使用 BLAST 直接在 NCBI 数据库中搜索产物。为什么保存这样的引物序列有用? 7. 使用 BLAST 搜索 "Primer pair 2" 的产物,将 Max target sequences 设置为最大,所有其他设置保持默认。你在数据库中找到多少个与你的产物匹配的命中?你能想象为什么 PrimerBLAST 指示你的引物对是特异性的吗? 8. PCR 不仅可以扩增基因组中的区域,还可以扩增 mRNA 的区域(mRNA 需要先转换为 cDNA)。执行此类 PCR 时,人们尝试设计跨越目标基因内含子的引物对(这也是 PrimerBLAST 中的一个选项)。你能推测为什么跨越内含子的引物可能有用吗?
项目准备练习
我们继续第一章的项目练习。 ARF5 和 IAA5 都属于拟南芥 (A. thaliana) 中的大家族。 现在专注于 ARF5 家族,通过识别同源物和寻找家族成员之间的保守部分来探索它。 在拟南芥内和该物种外执行此分析。 评估在哪些植物家族中检测到成员。
用几条要点分别描述以下项目。 你可以包含最多两个图表或表格。
- 材料与方法 你做了什么?你使用了哪些数据、数据库和工具?为什么选择它们?你选择了哪些重要设置?
- 结果 你发现了什么,主要结果是什么?报告相关数据、数字、表格/图表,并清楚地描述你的观察结果。
- 讨论与结论 结果合理吗?是否符合你的预期,还是你看到了令人惊讶的结果?结果意味着什么,你如何解释它们?不同的工具是否一致?你能得出什么结论?确保描述解释背后的预期和假设。
参考文献¶
{{ bibliography }}
术语表¶
仿射空位代价 (Affine gap costs): 区分空位开启和空位延伸代价的比对评分方案
BLAST: Basic Local Alignment Search Tool,基本局部比对搜索工具
BLOSUM: BLOck SUbstitution Matrix —— 一组蛋白质替换矩阵
一致序列 (Consensus sequence): 比对中最频繁出现的残基组成的序列
E 值 (E-value): 期望值 —— 在数据库中预期偶然出现具有观察得分或更高得分的命中数量(例如,使用 BLAST)
全局比对 (Global alignment): 比对策略,完整序列被比对
指导树 (Guide tree): 基于序列两两距离聚类的树,用于构建多序列比对
启发式算法 (Heuristic algorithm): 不保证找到最佳得分解的方法,而是采用通常能产生良好结果的经验法则
同源性 (Homology): 同源序列共享共同祖先
迭代优化 (Iterative refinement): 改善多序列比对的启发式方法
局部比对 (Local alignment): 比对策略,识别局部相似区域
模体 (Motif): 常见的序列模式
MSA: 多序列比对 (Multiple sequence alignment) —— 两条以上序列的比对
双序列比对 (Pairwise sequence alignment): 通过引入空位使两条序列比对以最大化得分的比对
PAM: Point Accepted Mutation —— 一组蛋白质替换矩阵
PCR: 聚合酶链式反应 (Polymerase chain reaction)
pHMM: 概隐马尔可夫模型 (profile hidden Markov model) —— 多序列比对的概率表示,允许在结构域数据库中搜索序列
引物 (Primers): PCR 中用于引发聚合酶的短单链 DNA 片段
渐进比对 (Progressive alignment): 基于指导树构建多序列比对的启发式方法
蛋白质一致性 (Protein identity): 双序列比对中相同氨基酸数量除以比对长度
蛋白质相似性 (Protein similarity): 双序列比对中相似和相同氨基酸数量之和除以比对长度
PSSM: 位置特异性评分矩阵 (Position Specific Scoring Matrix)
序列标志图 (Sequence logo): 比对的图形表示,显示该列中包含的信息
本文阅读量 次本站总访问量 次